We report the genomic analysis of thirteen SARS-CoV-2/hCOV-19 samples retrieved from patients in Scotland, in March 2020. Ten samples were processed and sequenced by the Royal Infirmary of Edinburgh in collaboration with the University of Edinburgh. Three samples were processed by the West of Scotland Specialist Virology Centre, NHSGGC and sent to the MRC-University of Glasgow Centre for Virus Research (CVR) for library preparation and sequencing. See our First report of COVID-19 in Scotland for details on the Oxford Nanopore Technologies MinION sequencing protocol utilised. The ARTIC Network V1 primers were used for sequencing.
The thirteen samples were named EDB003 to EDB013 and CVR06, CVR07 and CVR10, and have been deposited in GISAID (see table 1 for IDs and date of sampling).

To determine their relationship to circulating SARS-CoV-2/hCOV-19 variants, an evolutionary tree was inferred using these genomes and all complete human genomes available from GISAID; some GISAID sequences were excluded due to the presence of high numbers of mutations, presumably from sequence errors. Briefly, an alignment was generated using MAFFT and a maximum likelihood phylogenetic tree constructed using IQ-TREE with an HKY nucleotide substitution model and gamma rate heterogeneity model. The tree was visualised with FigTree and mid-point rooted.
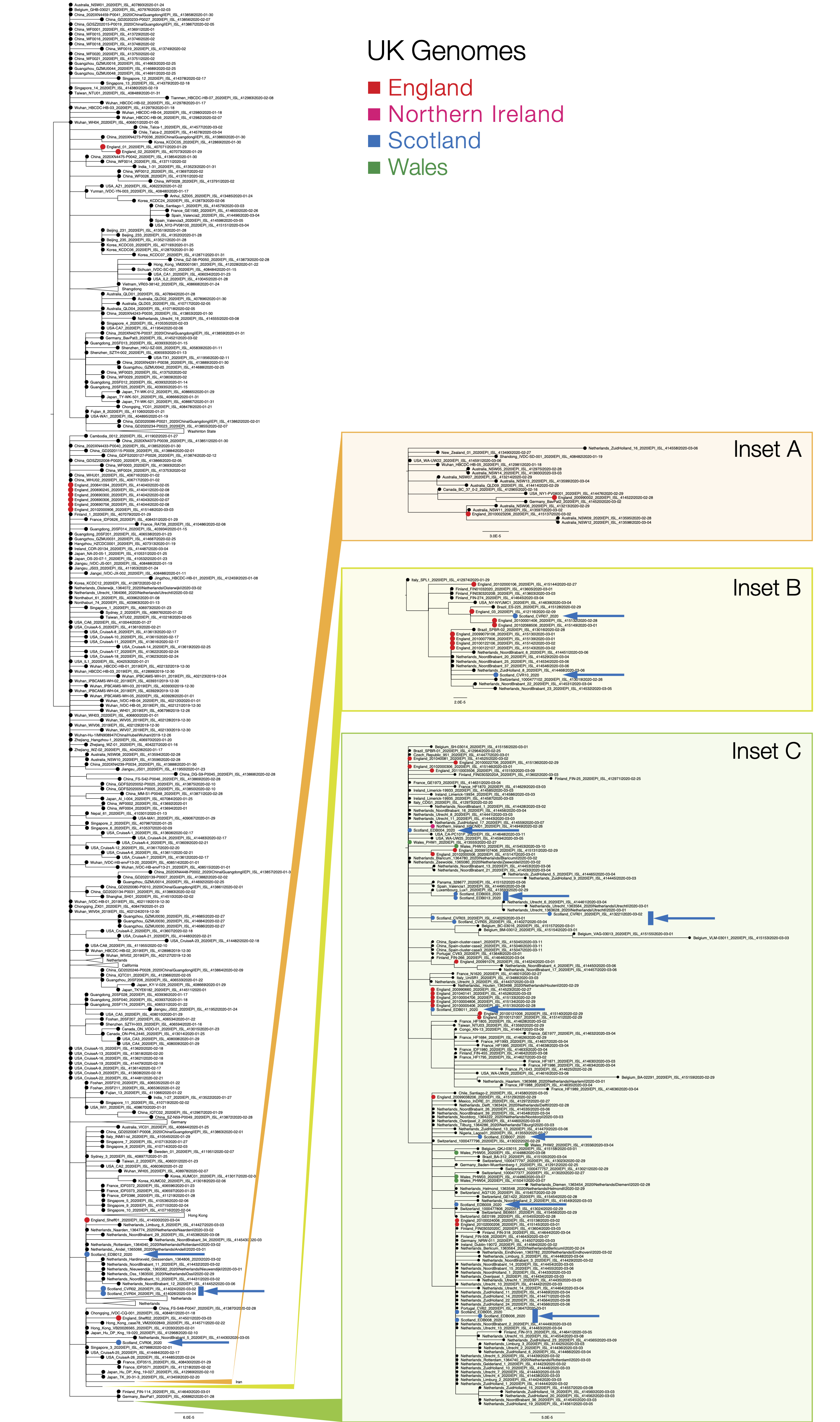
Figure 1 | Phylogenetic tree showing the relationship of CVR06, CVR07, CVR10 and EDB003 to EDB013 to other SARS-CoV-2/hCoV-19 genomes. Sequences from Scotland are in blue, England in red, Wales in green and Northern Ireland in magenta (see key). The countries indicated in the sequence names are where the individuals reside and not necessarily the origin of their infection, as most are recent travellers. The Insets show regions of the tree associated with widespread international circulation. Inset A is associated with the epidemic in Iran, Insets B & C show two established lineages that originate in Italy but have subsequently spread globally. The scale bar indicates substitutions per nucleotide site. PDF version: GISAID_ED_GLA_2020-03-17_574.iqtree.pdf (378.9 KB)
The phylogenetic analysis (Figure 1) indicates the majority of detected introductions of COVID-19 into Scotland are from returning travellers from Continental Europe, mostly Italy, whilst EDB011 is associated with an English cluster. Clustering is consistent with known travel histories. Introductions to Wales are all from Italy, while introductions to England mostly from Italy but are also from China and Iran.
Contributors (Edinburgh): RIE clinical researchers1: Martin McHugh, Rebecca Dewar, Kate Templeton; IGMM UoE2: Thomas Williams; IEB UoE3: Andrew Rambaut, Áine O’Toole
Contributors (Glasgow): CVR clinical researchers4: Natasha Jesudason, Kathy Li, Emma Thomson, Antonia Ho; CVR Genomics4: Kathy Smollett, Daniel Mair, Stephen Carmichael, Ana da Silva Filipe; CVR Bioinformatics4: Richard Orton, David L Robertson; WoSSVC, NHSGGC5: Alasdair MacLean, Rory Gunson.
1Royal Infirmary of Edinburgh; 2MRC Institute of Genetics and Molecular Medicine, University of Edinburgh; 3Institute of Evolutionary Biology, University of Edinburgh; 4MRC-University of Glasgow Centre for Virus Research (CVR); 5West of Scotland Specialist Virology Centre, NHSGGC and University of Edinburgh.
Acknowledgments
We would like to thank all the authors who have kindly deposited and shared genome data on GISAID. A table with genome sequence acknowledgments can be found here CoV-GLUE and below. The team in Edinburgh would also like to thank Mike Gallacher, Richard Kuo, Tim Regan and Amanda Warr (Roslin Institute, University of Edinburgh) for the kind loan of sequencing reagents.
Genome Data Acknowledgements
Table 2 | hCoV-2019 genome sequences used in this analysis, the GISAID accession numbers and submitting labs.