We updated our previous analysis of 93 genomes (Evolutionary & epidemiological analysis of 93 genomes) with 35 additional sequences.
Evolutionary analysis
We performed an evolutionary analysis to quantify the clock rate and TMRCA of COVID-19 sequences.
The method was identical to that presented in our original post.
We performed the analyses both on the original alignment and on a cluster-free subset of the alignment containing 86 sequences.
The conclusions of our original post still hold: at this stage, the clock rate and TMRCA estimates are sensitive to the tree prior.

Figure 1: Posterior distribution of clock rate, TMRCA, tree length (i.e. sum of branch
lengths), and tree divergence (= clock rate x tree length) for the 12 different tree
prior choices.
Epidemiological analysis
We performed an epidemiological analysis in which we fixed the clock rate and infectious period to estimate Re, the time of origin of the epidemic, and the overall number of cases. We performed the analyses both on the cluster-free alignment of 86 sequences and a subset of 41 sequences containing only samples taken outside of China. The method used is similar to that presented in our original post with a minor modification: we added a third time interval (after Feb 1st) to account for the reduced amount of available samples after that date in this dataset.
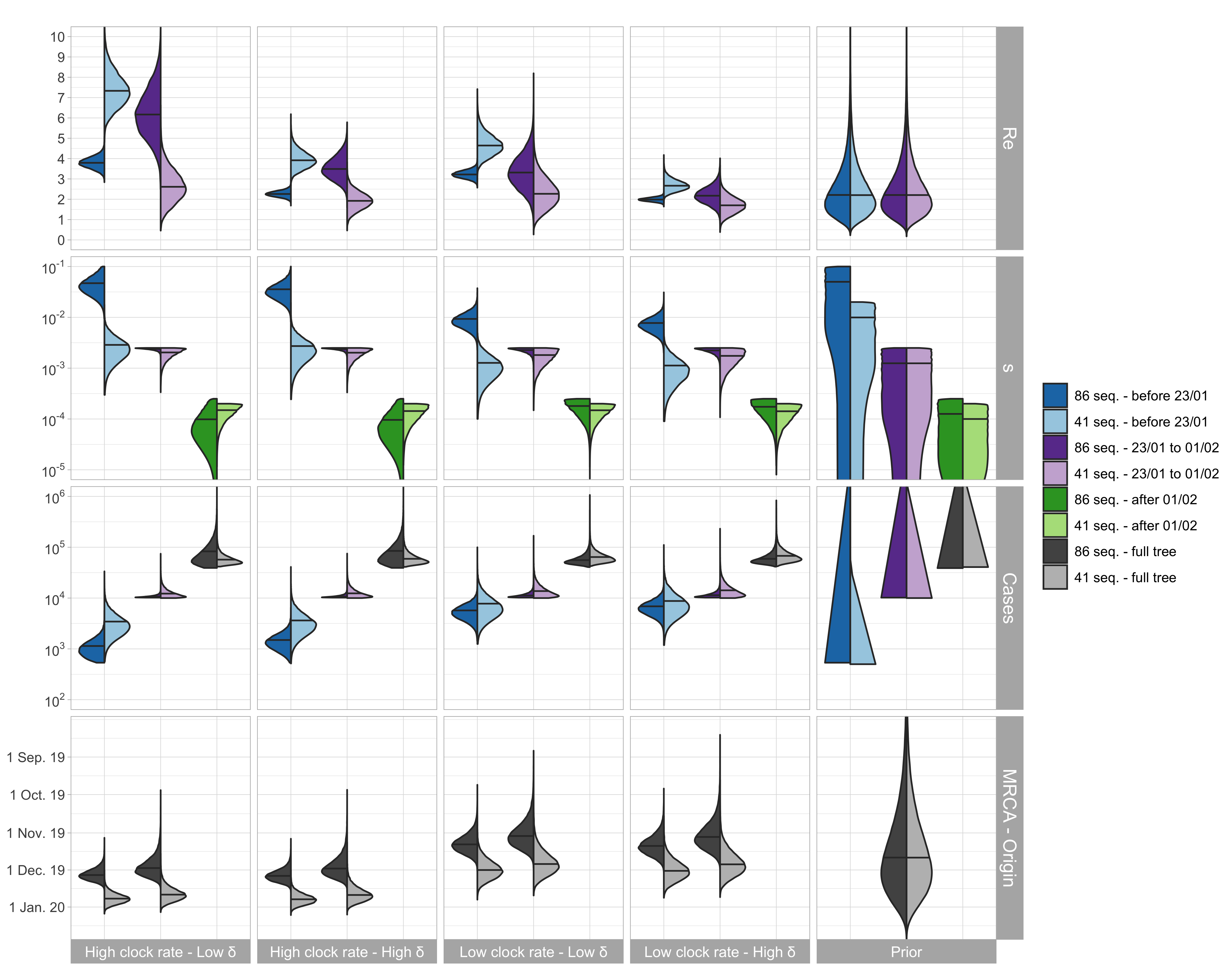
Figure 2: Posterior distribution of Re before Wuhan quarantine (which can be interpreted
as the R0), Re after Wuhan quarantine, sampling proportion s, total number
of cases (=#sequences / s), time of origin, and TMRCA for the cluster-free and the
outside-China alignment. In the bottom row, in each panel, we show on the left the
estimated TMRCA and on the right the estimated time of origin of the epidemic.
Caveats
Similar caveats to that mentioned in the original post apply for this update. In particular, the non-random mixing of the epidemic and the non-constant sampling effort are potential strong sources of bias of the estimates.
Acknowledegments
We thank Andrew Rambaut for his help and guidance with the processing of the GISAID sequences.
We gratefully acknowledge the Authors, the Originating and Submitting Laboratories for their sequence and metadata shared through GISAID, on which this research is based. Below is a full table containing the sequences used for the analysis.