SARS-CoV-2 reinfection by the new Variant of Concern (VOC) P.1 in Amazonas, Brazil
Felipe Naveca 1,2,3, Cristiano da Costa 2,4, Valdinete Nascimento 1,2,3, Victor Souza 1,2,3, André Corado 1,2,3, Fernanda Nascimento 1,2,3, Ágatha Costa 1,2,3, Débora Duarte 1,2,3, George Silva 1,2,3, Matilde Mejía 1,2,3, Karina Pessoa 1,2,3, Luciana Gonçalves 2,4, Maria Júlia Brandão 1,2,3, Michele Jesus 2,3,5, Rosemary Pinto 2,4, Marineide Silva 2,6, Tirza Mattos 2,6, Lígia Abdalla 7, João Hugo Santos 8, Rubens Costa-Filho 9, Gabriel Luz Wallau 3,10, Marilda Mendonça Siqueira 3,11, Edson Delatorre*, 3,12, Tiago Gräf*, 3,13, Gonzalo Bello*, 3,14, Paola Cristina Resende* ,3,11.
*These authors contributed equally to this work.
1 Laboratório de Ecologia de Doenças Transmissíveis na Amazônia, Instituto Leônidas e Maria Deane, Fiocruz, Manaus, Amazonas, Brazil.
2 Rede Genômica de Vigilância em Saúde do Estado do Amazonas, Manaus, Amazonas, Brazil.
3 Rede Genômica Fiocruz, Brazil.
4 Fundação de Vigilância em Saúde do Amazonas, Manaus, Amazonas, Brazil.
5 Laboratório de Diversidade Microbiana da Amazônia com Importância para a Saúde, Instituto Leônidas e Maria Deane, Fiocruz, Manaus, Amazonas, Brazil.
6 Laboratório Central de Saúde Pública do Amazonas, Manaus, Amazonas, Brazil.
7 Universidade do Estado do Amazonas, Manaus, AM, Brasil.
8 Hospital Adventista de Manaus, Manaus, AM, Brasil
9 Hospital Pró-Cardíaco, Rio de Janeiro, Brazil.
10 Departamento de Entomologia e Núcleo de Bioinformática, Instituto Aggeu Magalhães, Fiocruz, Recife, Pernambuco, Brazil.
11 Laboratório de Vírus Respiratórios e Sarampo, Instituto Oswaldo Cruz, Fiocruz, Rio de Janeiro, Brazil. SARS-CoV-2 National Reference Laboratory for the Brazilian Ministry of Health (MoH) and Reference Laboratory for the World Health Organization (WHO).
12 Departamento de Biologia. Centro de Ciências Exatas, Naturais e da Saúde, Universidade Federal do Espírito Santo, Alegre, Espírito Santo, Brazil.
13 Instituto Gonçalo Moniz, Fiocruz, Salvador, Bahia, Brazil.
14 Laboratório de AIDS e Imunologia Molecular, Instituto Oswaldo Cruz, Fiocruz, Rio de Janeiro, Brazil.
Summary
The SARS-CoV-2 lineage B.1.1.28 has been evolving in Brazil since February 2020, but the recent emergence of sub-lineages with convergent mutations in the spike (S) protein raises concern about the potential impact on viral infectivity and immune escape. The lineage P.1 (alias of B.1.1.28.1) is an emerging variant that harbours several amino acid mutations including S:K417T, S:E484K, and S:N501Y. This report describes the first confirmed case of reinfection with the P.1 lineage in a 29-years-old female resident in the Amazonas state, Brazil, previously infected with a B.1 lineage virus.
Keywords: COVID-19; SARS-CoV-2; reinfection; secondary infection; S:E484K; lineage B.1.1.28.P.1, Amazonas, Brazil
Introduction
Since the emergence of the coronavirus disease 2019 (COVID-19), a few cases of reinfection with phylogenetically distinct variants of SARS-CoV-2 have been reported (1). These reinfection cases might be the consequence of a limited and transitory protective immunity induced by the primo-infection or might reflect the reinfecting virus’s ability to evade the previous immune responses. The rapid spread in the United Kingdom (UK) and South Africa of emerging SARS-CoV-2 variants carrying several mutations in the receptor-binding domain (RBD) of the spike (S) protein (2,3) granting them the title of Variants of Concern (VOC). Among these mutations, E484K and N501Y are of particular concern since they potentially reduce antibody neutralization and increase affinity for ACE2 receptor (4-10). Of note, the first official record of a reinfection case with the emerging VOC B.1.1.7 circulating in the UK (11) was recently published.
The SARS-CoV-2 lineage B.1.1.28 has been circulating in Brazil since February 2020 without accumulating notable amino acid changes in the S protein (12,13). Nevertheless, recent genomic surveillance studies reported the emergence of two B.1.1.28 sub-clades with convergent mutations in the RBD of the S protein common to those detected in the UK and South African variants. One sub-clade, designated P.2 (alias for B.1.1.28.2), was first detected in Rio de Janeiro harboring the mutation S:E484K (14). The second sub-clade, designated P.1 (alias of B.1.1.28.1), was first detected in Japanese travelers returning from the Amazonas state (15) and due to the presence of several important mutations in the RBD (K417T, E484K, and N501Y) was also classified as a VOC. It is currently unclear to what extent the B.1.1.28 emerging lineages are disseminating in Brazil. However, two recent reports described the first documented cases of reinfection with the emerging P.2 lineage in individuals from the Brazilian Northeast region that were primo-infected by B.1.1.33 lineage variants (16,17).
In this study, we report the first documented case of reinfection with the newly emerging P.1 lineage in a 29-years-old individual from Amazonas, a Brazilian state that was severely hit by COVID-19 at the first epidemic wave between March and July, and is currently facing a spiraling surge of deaths since November 2020. The current case report here raises questions about the role of reinfections caused by this new VOC in the Amazonas’s second epidemic wave.
Case description
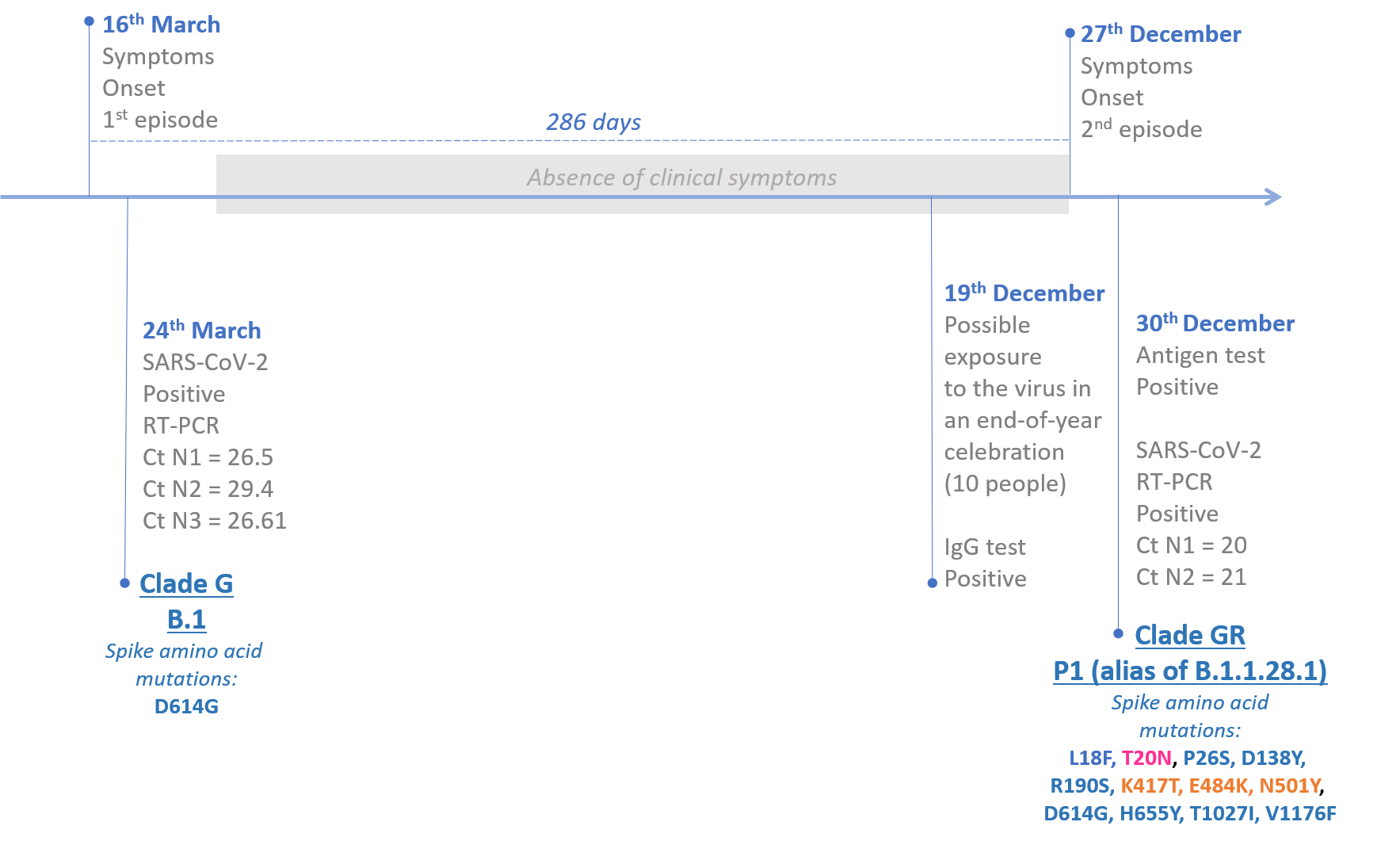
A 29-years-old woman resident in Manaus, Amazonas, Brazil, with no history of immunosuppression, presented two clinical episodes of COVID-19 infection with a gap interval of nine months (Figure 1). In the first episode on March 16th, the patient presented long-term fever and myalgia, cough, sore throat, nausea, and back pain. The patient was classified as having mild COVID-19 with no complications regarding these clinical manifestations and imaging exams. On December 19th, the patient reported participating in an end-of-year celebration with ten other people after testing positive in an IgG rapid test (Medlevensohn, RJ, Brazil). One of the meeting participants was RT-PCR confirmed for SARS-CoV-2 (18) infection on December 24th, and the patient exhibited the second symptomatic COVID-19 episode on December 27th, with fever, cough, sore throat, diarrhea, anosmia, ageusia, headache, runny nose, and resting pulse oximetry of 97%. Patient samples from the nasopharyngeal and pharyngeal swabs (NPS) were obtained on March 24th and December 30th, 2020. This study was approved by the UEA Ethics Committee CAAE: 25430719.6.0000.5016.
Figure 1. Timeline indicating the epidemiological, clinical, and laboratory events regarding the P.1 (alias of B.1.1.28.1) reinfection case in Manaus, Amazonas, Brazil. Mutation highlighted in pink indicates a new potential N-glycosylation site, and orange mutations indicate changes at the RBD.
Diagnostic Laboratory findings
The first and second NPS samples (e.g., March 24th and December 30th) were processed for diagnosis at Fiocruz Amazônia (Fiocruz/ILMD), which is part of the official network of the Ministry of Health for diagnostics and surveillance of the SARS-CoV-2 in the Amazonas state, Brazil. Briefly, total nucleic acid was extracted from the NPS specimens with Maxwell® RSC Viral Total Nucleic Acid Purification Kit (Promega, Madison, WI) and then immediately submitted to RT-qPCR detection of the SARS-CoV-2 RNA using the protocol developed by the US CDC, targeting the viral N gene, and human RNase P as the internal control (18). Both RT-PCR results were positive (mean Ct 27.5 - 1st sample; 20.5 - 2nd sample, Figure 1). Additionally, these samples were also positive for the DPP SARS-CoV-2 Antigen test system (Chembio Diagnostics, Medford, NY).
Genomic findings
We recovered high-quality SARS-CoV-2 whole-genomes from the two positive NPS samples from the suspected reinfection case (EPI_ISL_811148 and EPI_ISL_811149) and from other 10 subjects from Manaus that were also tested positive in December 2020 (EPI_ISL_833131 to EPI_ISL_833140). The whole-genome amplicons were generated as previously described (19) with a PCR scheme with nine overlapping amplicons (Supplemental File.pdf). NGS libraries were produced with Nextera XT and sequenced with MiSeq Reagent Micro Kit v2 (300-cycles). The FASTQ reads were obtained following the Illumina pipeline on BaseSpace, imported into Geneious v10.2.6, trimmed (BBDuk 37.25), and mapped (BBMap 37.25) against the reference sequence EPI_ISL_402124 available in EpiCoV database from GISAID (https://www.gisaid.org/). Consensus sequences with a mean coverage of 3,283x and 4,663x were generated for the first infection and reinfection case, respectively, excluding duplicate reads. Sequences were genotyped with the Pangolin software v2.1.7 (20) that indicate the presence of two different SARS-CoV-2 lineages in each COVID-19 episode: a B.1 lineage in the primo-infection and a P.1 lineage at reinfection.
To confirm the presence of two phylogenetically different SARS-CoV-2 lineages, sequences from the suspected reinfection case were aligned with all high quality (<2% of N) SARS-CoV-2 whole-genomes (>29 kb) from Brazil available in the EpiCoV database in GISAID by January 10th, 2021. Additionally, we also selected the four Japanese whole-genomes as the first representatives of the new P.1 lineage (EPI_ISL_792680 to EPI_ISL_792683). The ML phylogenetic analysis performed with IQ-TREE v2.1.2 (21) confirmed that the SARS-CoV-2 sequence from primo-infection branched in a small sub-clade together with three B.1 sequences from other Brazilian states (aLRT = 79%), while the sequence from the reinfection branched together with P.1 sequences isolated in Japan in 2021 along with five new sequences isolated in Manaus in December 2020 (aLRT = 100%) (Figure 2A). The P.1 reinfecting virus carried all 21 lineage-defining mutations (Supplementary Table 1) and displayed 11 amino acid changes in the S protein relative to the primo-infecting B.1 virus (Figure 1).
To further investigate the temporal scale of the novel P.1 lineage emergence and the viral strain associated with the reinfection case, we conducted a Bayesian analysis of all 10 SARS-CoV-2 sequences that branched within such clade. The time-scaled tree was estimated using a strict molecular clock model with a uniform substitution rate prior (8-10 ⨉ 10-4 substitutions/site/year), the HKY nucleotide substitution model, and the Bayesian skyline coalescent prior as implemented in BEAST 1.10 (22). Bayesian reconstructions traced the origin of the emerging P.1 lineage to December 1st (95% High Posterior Density [HPD]: November 15th – December 4th) and the most recent common ancestor of the reinfecting virus and the closest P.1 viruses to December 23th (95% HPD: December 12th – December 29th) (Figure 2B). This time frame confidently excludes the possibility of long-term persistence of the P.1 virus since primo-infection and traced the second infection a few days earlier before the onset of reinfection symptoms (December 27th).
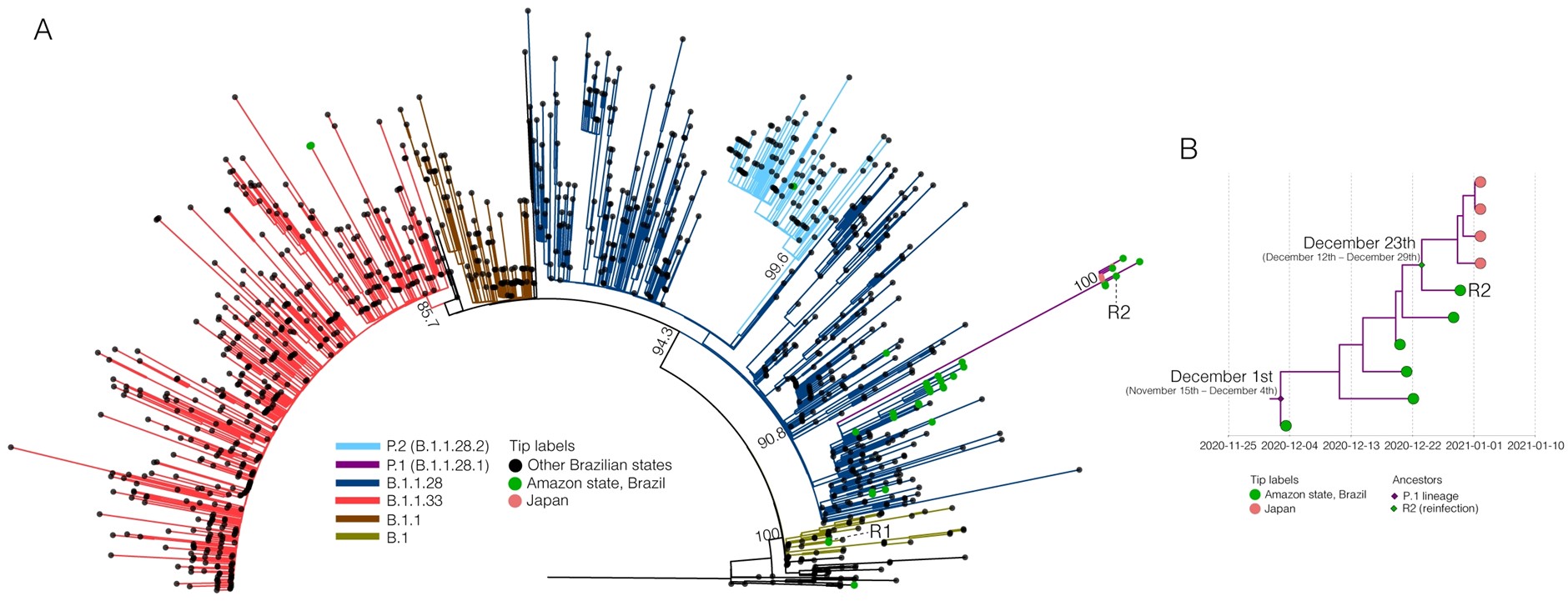
Figure 2. Evolutionary analysis of the SARS-CoV-2 genomic sequences from the reinfection case. A. ML phylogenetic tree of all SARS-CoV-2 whole-genome sequences from Brazil. Branches and tips were colored based on the lineage and sampling location, respectively, following the legend’s color code. Branch supports (aLRT) are presented at key nodes. The first (R1) and the second (R2) genomes obtained from the reinfection case are indicated. The scale of the phylogenetic branches is given as substitutions per nucleotide site. B. Time-scaled Bayesian MCC tree of SARS-CoV-2 P.1 lineage. Tips are colored according to the sampling location. Colored diamonds indicate the key ancestral nodes representing the MRCA of the P.1 clade (green) and the MRCA of the reinfecting virus, and the closest P.1 sequences (purple).
Discussion
This study described the first case of reinfection with the emerging P.1 lineage (carrying mutations S:K417T, S:E484K and S:N501Y) in a young, immunocompetent woman infected with a B.1 lineage virus nine months before. Notably, the patient had equal moderate symptomatic infections during both episodes and higher viral load (SARS-CoV-2 RT-PCR Ct value) in nasopharyngeal and pharyngeal samples obtained at reinfection compared with those from primo-infection. Although samples at primo-infection were collected later relative to symptom onset compared with samples at reinfection, these findings support that natural infections do not necessarily prevent reinfection or mitigate disease at subsequent infections (11,23). Furthermore, the low Ct values (< 25) detected at reinfection suggests that the subject may have been infectious and contributed to the onward transmission of the virus in the population (24).
A recent longitudinal study in health care workers suggests that post-infection anti-SARS-CoV-2 IgG antibodies are associated with protection from reinfection for most people for at least six months (25). We may speculate that the patient here described developed a transient protective immunity after primo-infection, but the anti-SARS-CoV-2 antibodies substantially decayed by the time of reinfection nine months later (26). However, the positive IgG rapid test obtained only eight days before the onset of symptoms in the second episode suggests that reinfection probably occurred in the face of pre-existing anti-SARS-CoV-2 antibodies. Another hypothesis is that the analyzed COVID-19 convalescent individual produced total IgG antibodies with low neutralizing potency and was thus susceptible to reinfection with new viral variants (27). Finally, the cases of reinfection with the B.1.1.28-derived emerging lineages (i.e P.1 and P.2) detected here and in previous reports (16,17) might also reflect the ability of S:K484 viruses to escape from anti-SARS-CoV-2 neutralizing antibodies induced during primo-infection with S:E484 variants that predominated during the “first-wave” epidemic in Brazil (4-7).
Urgent studies are necessary to determine whether reinfection with newly emerging lineages harboring the mutation S:E484K is a widespread phenomenon or is limited to a few sporadic cases. It will also be crucial to understand the extent to which reinfection contributes to the forward transmission of SARS-CoV-2 in previously exposed populations and the rising number of SARS-CoV-2 cases observed in Amazonas and other Brazilian states during December 2020 - January 2021.
Acknowledgment
We would like to thank the funding support from CGLab/MoH (General Laboratories Coordination of Brazilian Ministry of Health), CVSLR/FIOCRUZ (Coordination of Health Surveillance and Reference Laboratories of Oswaldo Cruz Foundation), CNPq COVID-19 MCTI 402457/2020-0 and 403276/2020-9; INOVA Fiocruz VPPCB-005-FIO-20-2 and VPPCB-007-FIO-18-2-30; FAPERJ: E26/210.196/2020; FAPEAM (PCTI-EmergeSaude/AM call 005/2020 and Rede Genomica de Vigilancia em Saude - REGESAM). We also appreciate the support of all Genomic Coronavirus Fiocruz Network members, the Multi-user Research Facility of Biosafety Level 3 Platform of the Oswaldo Cruz Institute (IOC), Fiocruz, and the CGLab/MoH and Secretary of Surveillance and Health of the Brazilian MoH (SVS-MS). Additionally, we are thankful for all that contribute to the EpiCoV database from the GISAID initiative with high-quality SARS-CoV-2 genomes. All genomes used in this study are described in the acknowledgment Supplementary Table 1 (Suplementary_table 1_acknowledgement_table_GISAID.pdf).
References
1. Babiker A, Marvil C, Waggoner JJ, Collins M, Piantadosi A. The Importance and Challenges of Identifying SARS-CoV-2 Reinfections. J Clin Microbiol 2020.
2. Rambaut AL, N.; Pybus, O.; Barclay, W.; Barrett, J.; Carabelli, A.; Connor, T.; Peacock, T.; Robertson, D.; Volz, E.; on behalf of COVID-19 Genomics Consortium UK (CoG-UK); . Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. Virologicalorg. https://pando.tools/2020.
3. Tegally H, Wilkinson E, Giovanetti M, et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv 2020.
4. Baum A, Fulton BO, Wloga E, et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020;369:1014-8.
5. Greaney AJ, Loes AN, Crawford KHD, et al. Comprehensive mapping of mutations to the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human serum antibodies. bioRxiv 2021.
6. Liu Z, VanBlargan LA, Rothlauf PW, et al. Landscape analysis of escape variants identifies SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. bioRxiv 2020.
7. Weisblum Y, Schmidt F, Zhang F, et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. Elife 2020;9.
8. Andreano E, Piccini G, Licastro D, et al. SARS-CoV-2 escape in vitro from a highly neutralizing COVID-19 convalescent plasma. bioRxiv 2020.
9. Zahradník J, Marciano S, Shemesh M, et al. SARS-CoV-2 RBD in vitro evolution follows contagious mutation spread, yet generates an able infection inhibitor. bioRxiv 2021.
10. Nelson G, Buzko O, Spilman P, Niazi K, Rabizadeh S, Soon-Shiong P. Molecular dynamic simulation reveals E484K mutation enhances spike RBD-ACE2 affinity and the combination of E484K, K417N and N501Y mutations (501Y.V2 variant) induces conformational change greater than N501Y mutant alone, potentially resulting in an escape mutant. bioRxiv 2021.
11. Harrington D, Kele B, Pereira S, et al. Confirmed Reinfection with SARS-CoV-2 Variant VOC-202012/01. Clin Infect Dis 2021.
12. Candido DS, Claro IM, de Jesus JG, et al. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science 2020;369:1255-60.
13. Resende P. C. D, E., Gräf T., Mir D., Motta F.C., Appolinario L., Paixão A. C., Mendonça A. C., Ogrzewalska M., Caetano B., Wallau G. L., Docena C., Santos M. C., Ferreirra J., Sousa Junior E., Silva S., Fernandes S., Vianna L. A., Souza L., Ferro J. F, Nardy V., Santos C., Riediger I., Debur M., Croda J., Oliveira, W, Abreu A, Bello G… Siqueira M. M. Evolutionary dynamics and dissemination pattern of the SARS-CoV-2 lineage B.1.1.33 during the early pandemic phase in Brazil. Frontier in Microbiology 2020.
14. Voloch CM, Silva F Rd, de Almeida LGP, et al. Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. medRxiv 2020.
15. Japan.; NIoID. ブラジルからの帰国者から検出された新型コロナウイルスの新規変異株について. Coronavirus disease (COVID-19)2021:4.
16. Resende PCB, J.F.; Vasconcelos, R.H.T.; Arantes I.; Appolinario L.; Mendonça, A.C.; Paixao, A.C.; Rodrigues A.C.; Silva, T.; Rocha, A.S.; Pauvolid-Corrêa, A.; Motta, F.C.; Teixeira, D.L.F.T.; Carneiro, T.F.O.; Freire Neto, F.P.F.; Herbster, I.D.; Leite, A.B.; Riediger, I.N.; Debur, M.C.; Naveca, F.G.; Almeida, W.; Livorati, M.; Bello, G.; Siqueira, M.M. Spike E484K mutation in the first SARS-CoV-2 reinfection case confirmed in Brazil, 2020. Virologicalorg2020.
17. Vasques Nonaka CKMF, M.; Gräf, T.; Almeida Mendes, A.V.; Santana de Aguiar, R.; Giovanetti, M.; Solano de Freitas Souza, B. Genomic Evidence of a Sars-Cov-2 Reinfection Case With E484K Spike Mutation in Brazil. Preprints 2021.
18. Centers for Disease Control and Prevention. CDC 2019-Novel Coronavirus (2019-nCoV) Real-Time RT-PCR Diagnostic Panel. 2020:80.
19. Nascimento VAD, Corado ALG, Nascimento FOD, et al. Genomic and phylogenetic characterisation of an imported case of SARS-CoV-2 in Amazonas State, Brazil. Mem Inst Oswaldo Cruz 2020;115:e200310.
20. Rambaut A, Holmes EC, O’Toole A, et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol 2020.
21. Minh BQ, Schmidt HA, Chernomor O, et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol 2020;37:1530-4.
22. Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol 2018;4:vey016.
23. Tillett RL, Sevinsky JR, Hartley PD, et al. Genomic evidence for reinfection with SARS-CoV-2: a case study. Lancet Infect Dis 2021;21:52-8.
24. Bullard J, Dust K, Funk D, et al. Predicting infectious SARS-CoV-2 from diagnostic samples. Clin Infect Dis 2020.
25. Lumley SF, O’Donnell D, Stoesser NE, et al. Antibody Status and Incidence of SARS-CoV-2 Infection in Health Care Workers. N Engl J Med 2020.
26. Selhorst P, Van Ierssel S, Michiels J, et al. Symptomatic SARS-CoV-2 reinfection of a health care worker in a Belgian nosocomial outbreak despite primary neutralizing antibody response. Clin Infect Dis 2020.
27. Robbiani DF, Gaebler C, Muecksch F, et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature 2020;584:437-42.