SARS-CoV-2 lineage assignment is more stable with UShER
Adriano de Bernardi Schneider1,2,![]() ,*, Michelle Su3,
,*, Michelle Su3,![]() ,Angie Hinrichs1,2, Jade Wang3, Helly Amin3, John Bell4, Debra A. Wadford4, Áine O’Toole5, Emily Scher5, Marc D. Perry 1, Rachel Colquhoun5, Yatish Turakhia6, Scott Hughes3, Russ Corbett-Detig1,2
,Angie Hinrichs1,2, Jade Wang3, Helly Amin3, John Bell4, Debra A. Wadford4, Áine O’Toole5, Emily Scher5, Marc D. Perry 1, Rachel Colquhoun5, Yatish Turakhia6, Scott Hughes3, Russ Corbett-Detig1,2
1 Genomics Institute, University of California Santa Cruz, Santa Cruz, CA, USA
2 Department of Biomolecular Engineering, University of California Santa Cruz, Santa Cruz, CA, USA
3 New York City Department of Health and Mental Hygiene, New York, NY, USA
4 California Department of Public Health (CDPH), VRDL/COVIDNet, Richmond, CA 94804, USA
5 Institute of Evolutionary Biology, University of Edinburgh, Edinburgh EH93FL, UK
6 Department of Electrical and Computer Engineering, University of California San Diego, San Diego, CA 92093, USA
![]() These authors contributed equally to this work.
These authors contributed equally to this work.
*Corresponding author: [email protected]
Introduction
The assignment of Pango lineages (1,2) to newly sequenced SARS-CoV-2 genomes has been central in aiding health officials to trace the spread of the virus locally and globally and identifying differences among viral lineages (3). Currently, the most commonly used tool for the assignment of newly isolated SARS-CoV-2 genomes to pango lineages is pangolin, which offers pangoLEARN (the default) and UShER modes (4,5,6). PangoLEARN aims for a rapid assignment of lineages using a machine learning algorithm. UShER performs a phylogenetic placement to identify the lineage corresponding to a newly sequenced genome. PangoLEARN is substantially faster than UShER. However, because the pango lineage nomenclature system is phylogenetically derived (1), it is possible that UShER is more accurate and stable in lineage assignments across subsequent releases. Given the epidemiological importance of assigning strains the correct lineages, here, we sought to evaluate the consistency and accuracy of the two main methods currently available.
The Pango lineage system is explicitly designed to be updated with the pandemic as the virus continues to evolve (1). For that reason, some lineage reassignments are expected for a given genome sequence. For example, a sample originally designated in one lineage might be reassigned to a new daughter lineage of its original assignment. This occurs as an expected part of the Pango system when new lineages are designated. However, in some cases, a sample may be reassigned to a non-descendant lineage in subsequent versions of pangoLEARN or UShER due to error in the assignment approach. Hereafter, we refer to such lineage reassignments as non-permitted lineage changes. Instability in lineage assignments might cause problems for interpretation that relies on precise and reliable lineage definitions for individual samples.
Note that pangolin also incorporates another lineage assignment approach, Scorpio, which takes a set of lineage-defining “constellation” datasets with rules to classify each sequence by its specific mutations. Scorpio requires a manually curated constellation definition for each assignable lineage; typically, curation resources are limited to Variants of Concern, Variants of Interest or Variants Under Monitoring (https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/). Scorpio is therefore applicable only to a subset of the SARS-CoV-2 lineages and is not evaluated further here. When pangoLEARN or UShER makes an assignment that conflicts with Scorpio’s assignment, pangolin overrides the pangoLEARN or UShER assignment with the Scorpio assignment.
Methods
We evaluated the consistency of lineage assignment using pangoLEARN and UShER on two datasets. The first is 7229 genomes sampled in New York City by DOHMH PHL, NYC Pandemic Response Lab, and contributors to the GISAID Initiative (Supplemental Table 1) (7). The second is a set of 20198 genomes sampled in California via the CDPH COVIDNet sequencing effort. These analyses were done largely independently but arrive at the same qualitative conclusions.
New York City sequence analysis
We evaluated the assignment of 7229 Delta lineage SARS-CoV-2 from New York City at seven timepoints between September 22 to November 4, 2021, spanning Pango designation v.1.2.76-91, pangolin v.3.1.11-16, and pangoLEARN 2021-09-17 to 2021-10-18. The three primary metrics used to assess results were monophyly of assigned lineages, stability of lineage calls over time, and incidence of lineage calls overwritten by Scorpio. Sample consensus sequences were filtered to have >= 97% genome coverage and used to build a phylogenetic tree using MAFFT (8) and IQ-TREE (9). To aid in the visualization of results, this tree was downsampled iteratively by collapsing tips that had the same lineage call at all timepoints into one datapoint. The final tree was downsampled via this method six times and had a final tip count of 853.
California sequence analysis
We evaluated the assignment of 20198 SARS-CoV-2 sequences isolated in the state of California from the California Department of Public Health across four versions of pangoLEARN and UShER spanning Pango designation v1.2.76-93, pangolin v.3.1.13-16, and pangoLEARN releases (which also include phylogenetic trees used by UShER) 2021-09-17 - 2021-11-09. We performed a Pearson’s product-moment correlation between genome coverage and number of lineage calls across all versions of each method and number of non-permitted lineage changes. We also evaluated the correlation between sample parsimony scores (i.e., number of private mutations when the sample is placed on the tree) and number of lineage calls. We do this because the sample parsimony scores can serve as a proxy for sequencing quality. That is, samples with many private mutations (higher parsimony scores) will typically be lower quality genome assemblies. We created a list of expected versus non-permitted lineage assignments between each version based on the number of newly designated lineages. This was used to create a comparison between all the versions which was then visualized in a Sankey diagram for each assignment method. Finally, we evaluated the distribution of samples based on genome completeness above a 90% threshold across the total number of calls (lineage assignments) and number of non-permitted lineage assignments.
Results & Discussion
New York City analyses
PangoLEARN and pUShER lineage assignments have a high level of concordance in the New York City dataset. However, at the beginning and end of the study period, pangoLEARN had difficulty calling newly some of the newly identified lineages by reclassifying many samples across the phylogenetic tree into a sublineage with no distinct clustering and artificially inflating its prevalence: AY.4 in September and AY.43 in November (Figure 1 and 2). It also appears to be more prone to outliers than pUShER whereby otherwise monophyletic clades were punctuated by other lineage calls. pangoLEARN performance on calling AY.4 improved with time as can be seen by its exclusion from Figure 2 as it was no longer being overcalled.
We then investigated the likelihood of lineage reassignments by either model (Figure 3). In the case of pangoLEARN, there is a substantial proportion of the downsampled tree samples that undergo lineage reassignment as compared to UShER. As sublineages of Delta were being quickly defined, there is an expectation that there are lineage reassignments that are appropriate. In these cases, one would expect two lineage assignments and generally from Delta to a sublineage and not a sublineage to sublineage change. This is not the case with most pangoLEARN reassignments which can likely be explained with the aforementioned issue of overcalling AY.4 or other newly emerging lineages. In contrast, most lineage reassignments by UShER are stable changes from Delta to a sublineage. Further, the exhibited clustering of these changes are expected when a monophyletic clade is reassigned.
While scorpio is not generally applicable to lineage assignments of many SARS-CoV-2 sequences, it can be used as a stopgap for accurately assigning newly defined lineages. In our full sample set of 7229 Delta sequences, scorpio was used only in a minority of samples. In the case of pangoLEARN, 207 samples had lineage calls overwritten by scorpio at least once compared to 36 samples by UShER. Of note, one of the most commonly targeted lineage calls for pangoLEARN was AY.4.
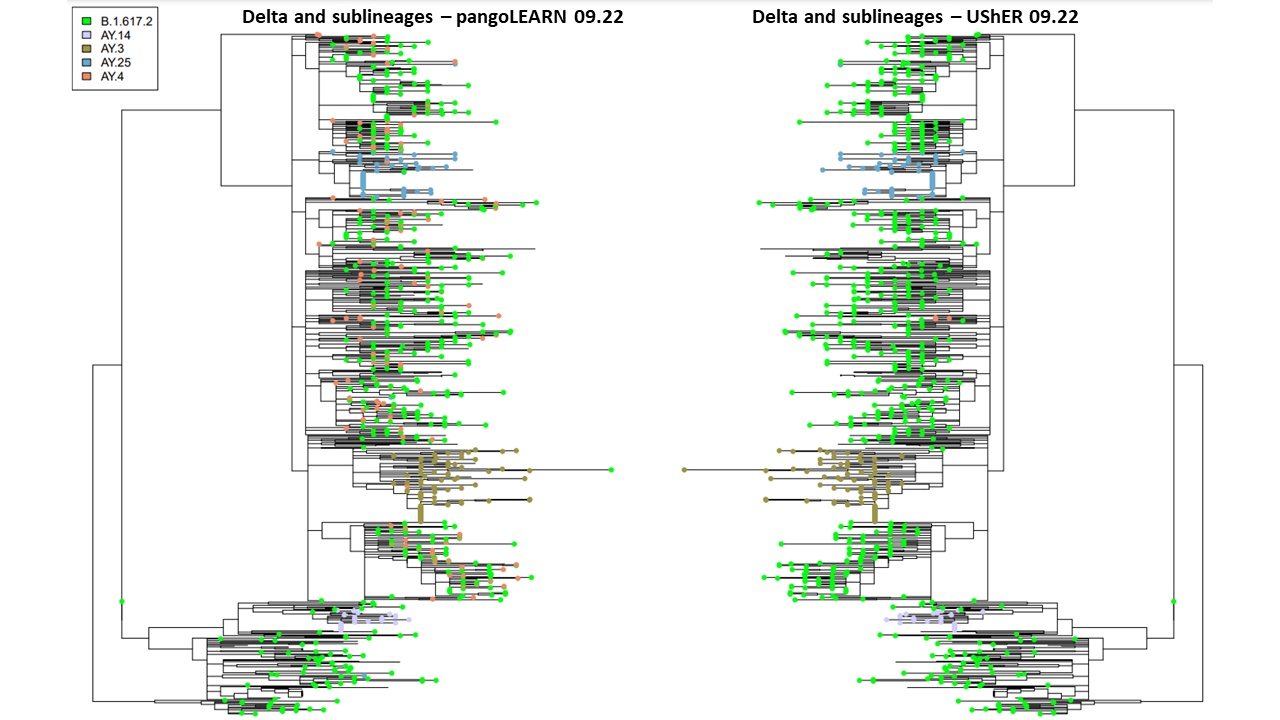
Figure 1. Comparison of pangoLEARN and UShER on 09.22.2021 on downsampled phylogenetic tree. 853 samples are represented but only top five lineages are designated by tip labels.
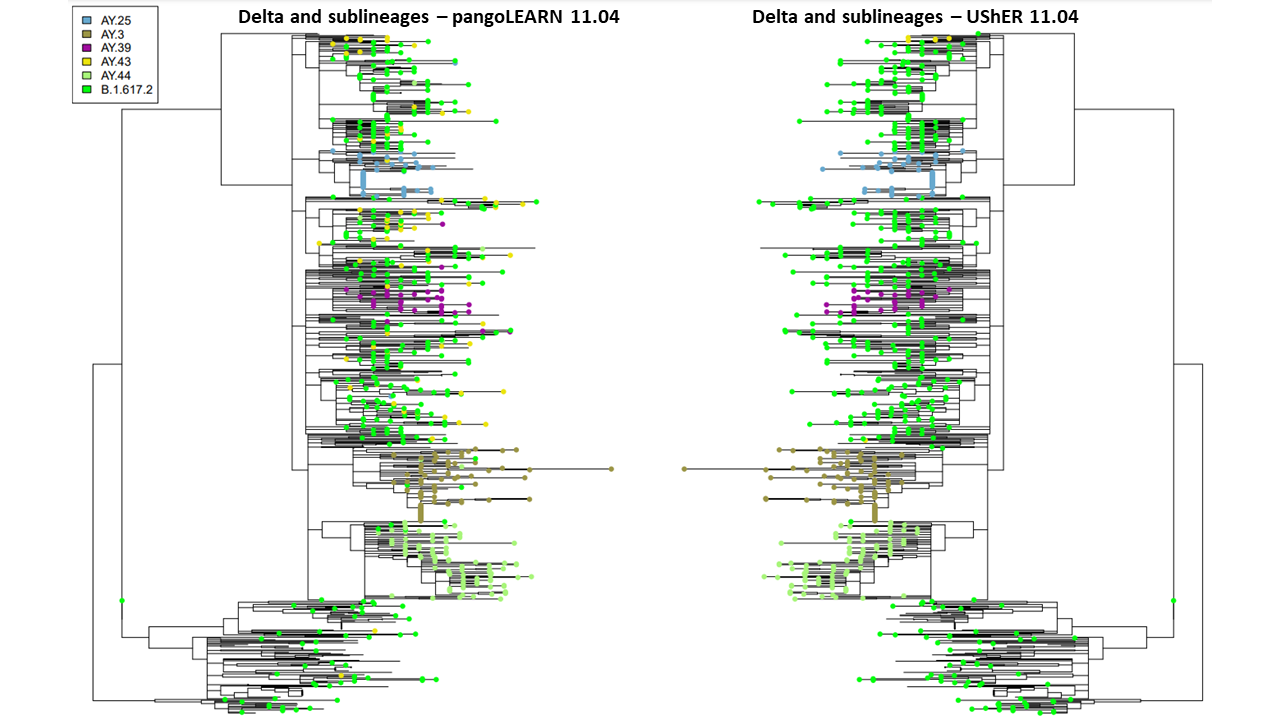
Figure 2. Comparison of pangoLEARN and UShER on 11.04.2021 on downsampled phylogenetic tree. 853 samples are represented but only top six lineages are designated by tip labels.
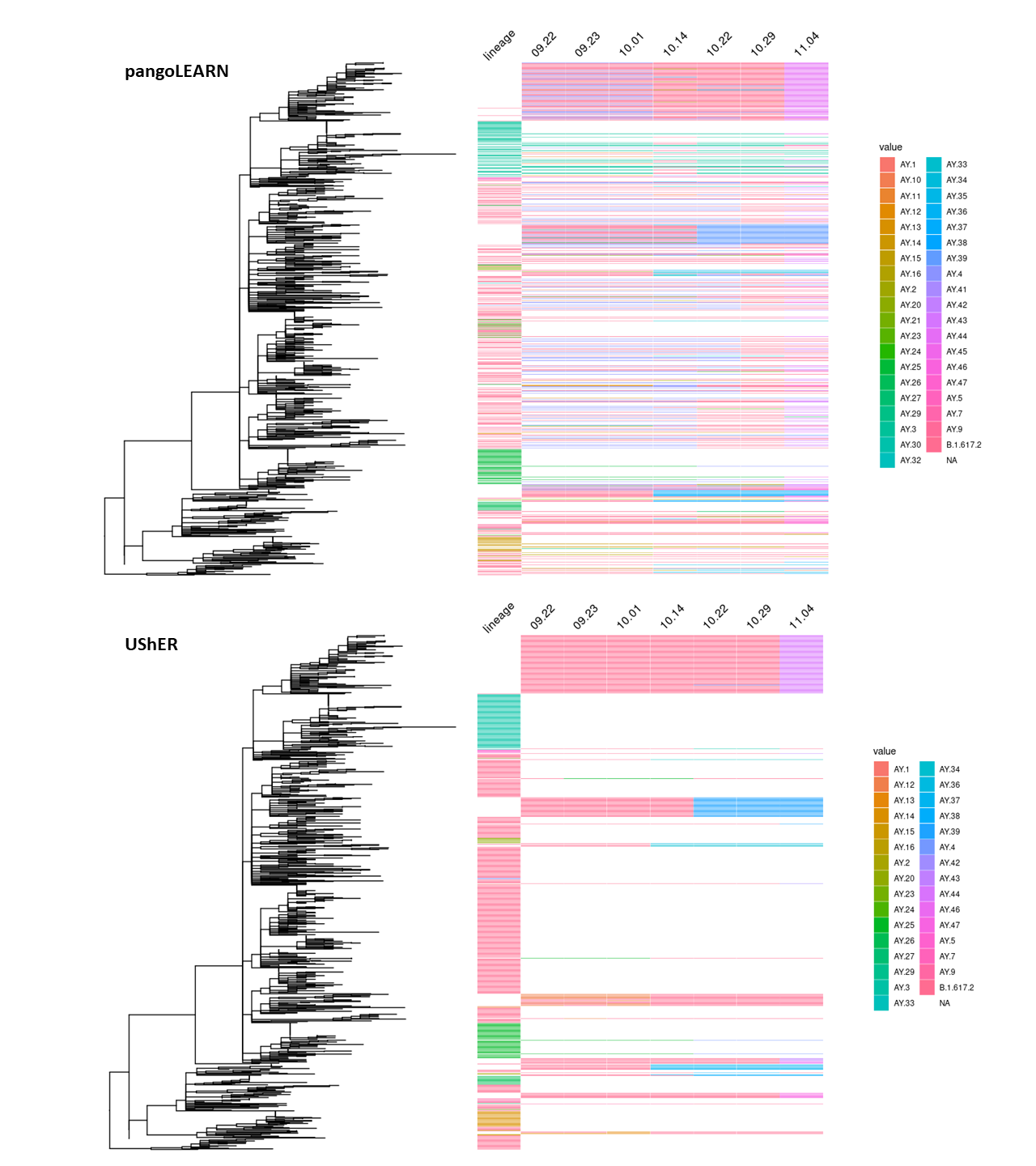
Figure 3. pangoLEARN and UShER lineage stability between 09.22.21 and 11.04.21 on downsampled tree. 853 samples are represented and lineage calls are designated to the right of the tree. The lineage column is populated only if the sequence’s lineage assignment did not change during the study period. Otherwise, the lineage calls are indicated under their respective dates.
California analyses
Within the dataset from California, UShER presented more stability in terms of valid lineage assignments across all four versions in comparison to pangoLEARN (Figure 4 and 5). The total number of single and double lineage assignments made by UShER is similar to pangoLEARN over the four versions analyzed (Table 1). However, UShER outperformed pangoLEARN when looking at the number of non-permitted lineage changes. UShER assignments for 97.63% of sequences were consistent (zero non-permitted lineage changes) across all four versions, whereas pangoLEARN assigned 78.74% of the sequences with zero non-permitted lineage calls in subsequent versions (Table 2). We emphasize that this is an approximation for consistency of each method, and does not necessarily indicate that the UShER calls are more accurate. It is possible that UShER is consistently wrong. However, we think that UShER is probably more accurate.
We further investigated the possible reasons behind the unstable lineage assignments. Genome coverage was a clear contributing factor to changes in lineage assignments by both applications. While pangoLEARN had a maximum of four lineage assignments for a single sequence, UShER had a maximum of three, indicating that some assignments toggled between states (e.g., None > B.1.617.2 > AY.44 > B.1.617.2 across three subsequent rounds of assignment) (Figure 6A). Sequences with reference genome coverage above 97.5% were generally assigned consistently with UShER (Figure 6B). pangoLEARN appears to perform well with sequences that present high reference genome coverage (>98.75%), but presents more unexpected calls when below this threshold (Figures 6C and 6D).
The correlation between genome coverage and number of non-permitted lineage assignments was stronger with pangoLEARN (r = -0.38) than with UShER (r = -0.16). The correlation between genome coverage and number of calls was stronger with pangoLEARN (r = -0.21) compared with UShER (r = -0.02, no correlation). There was no correlation between the number of non-permitted lineage assignments and parsimony score for both UShER (r = 0.08) and pangoLEARN (r = 0.04). These results indicate that pangoLEARN is more susceptible to reference genome coverage in comparison to UShER and parsimony score is not a contributing factor to the unexpected calls by both applications.
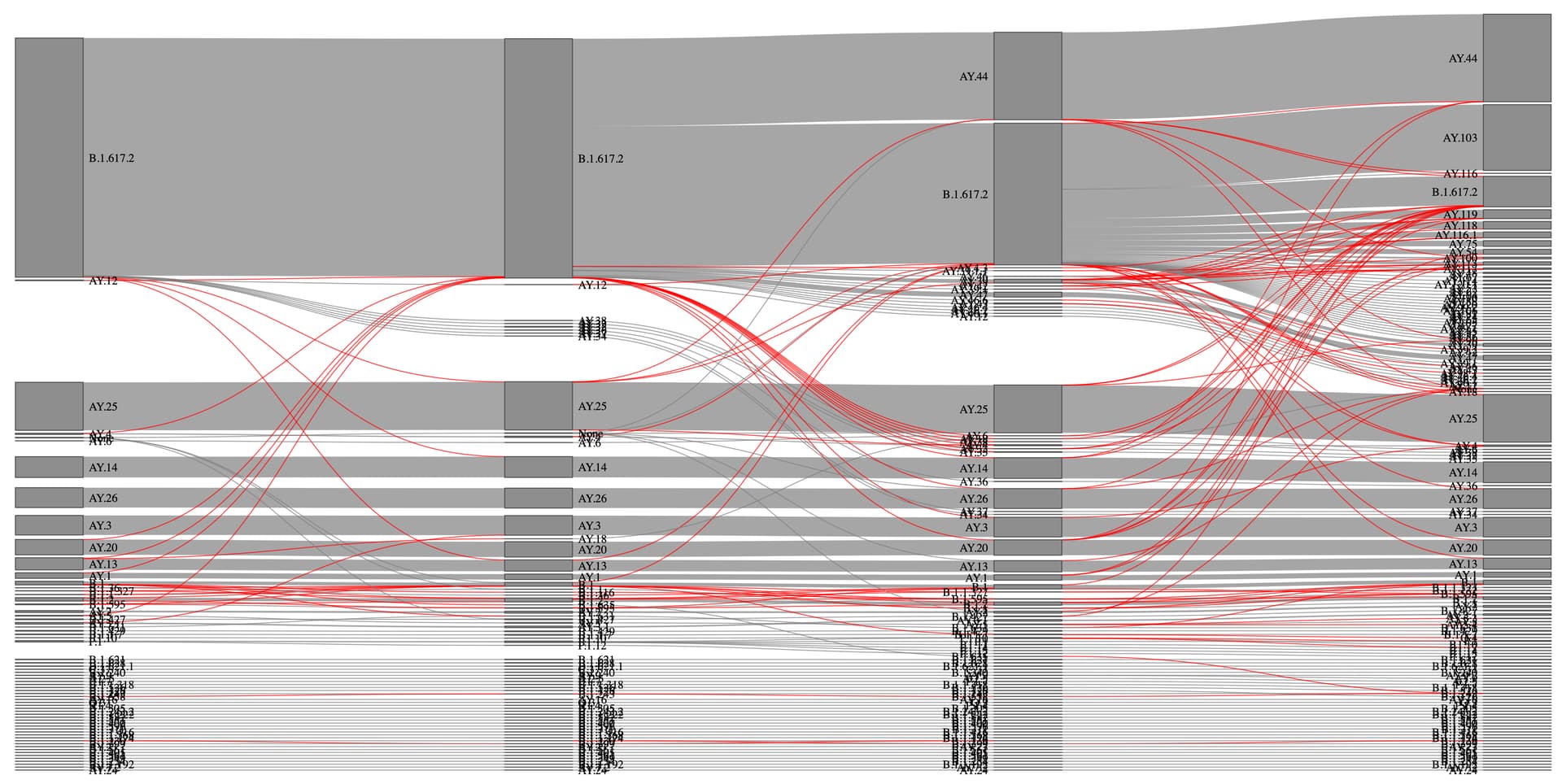
Figure 4. Sankey diagram of lineage assignment using UShER across four different versions. Red lines represent unexpected changes between versions.
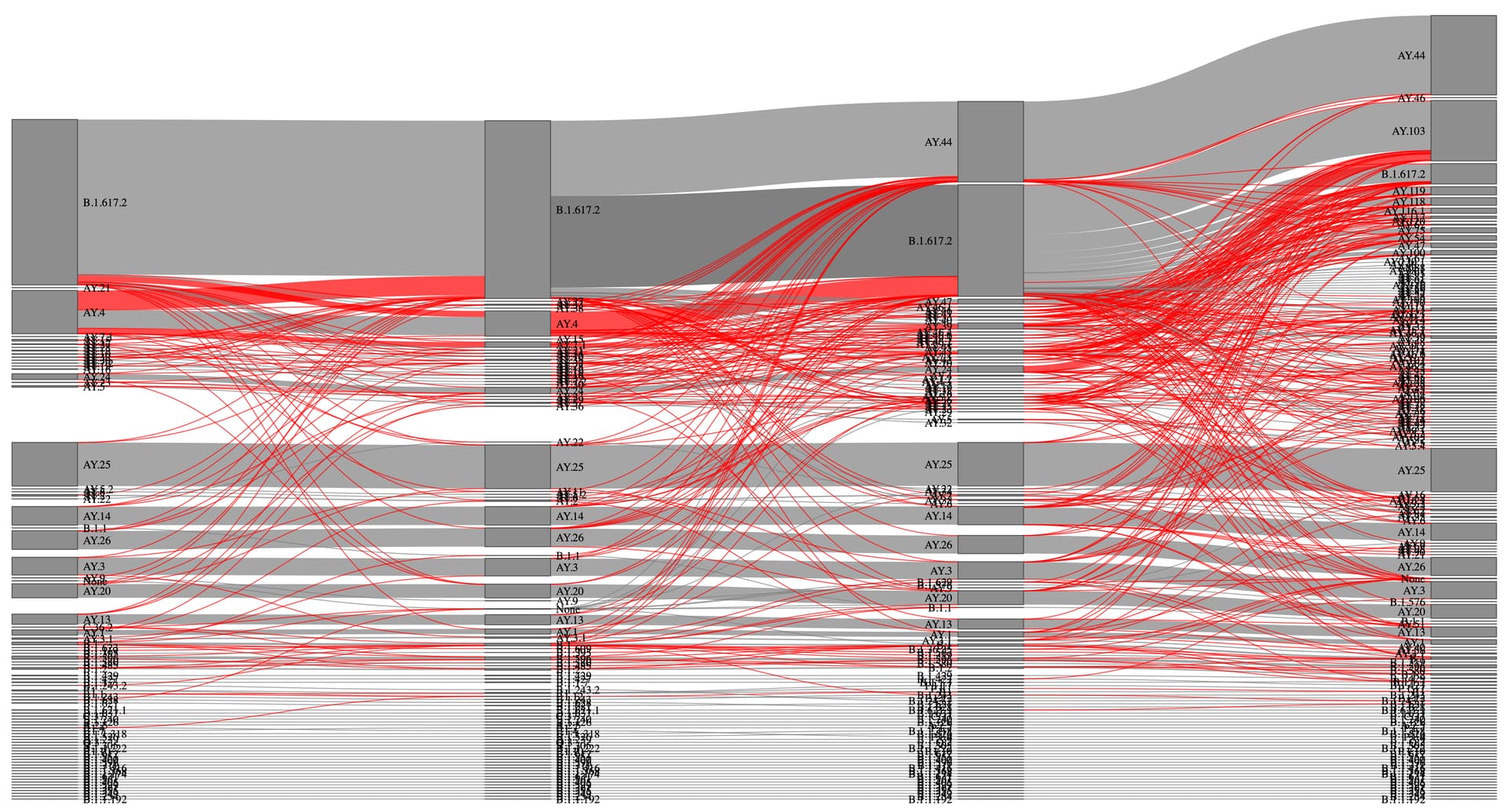
Figure 5. Sankey diagram of lineage assignment using pangoLEARN across four different versions (v.13,14,15 and 16). Red lines represent unexpected changes between versions.
Table 1. Number of lineage assignments using pangoLEARN and pUShER across four different versions (v. 13,14,15 and 16).
| application\n of calls | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| pangoLEARN | 8490 (42%) | 8502 (42.1%) | 2900 (14.4%) | 306 (1.5%) |
| pUShER | 9133 (45.22%) | 10963 (54.28%) | 102 (0.05%) | 0 |
Table 2. Number of non-permitted lineage changes using pangoLEARN and pUShER across four different versions (v. 13,14,15 and 16).
| application/non-permitted changes | 0 | 1 | 2 | 3 |
|---|---|---|---|---|
| pangoLEARN | 15904 (78.74%) | 3088 (15.29%) | 928 (4.59%) | 278 (1.38%) |
| pUShER | 19719 (97.63%) | 367 (1.82%) | 100 (0.5%) | 12 (0.05%) |
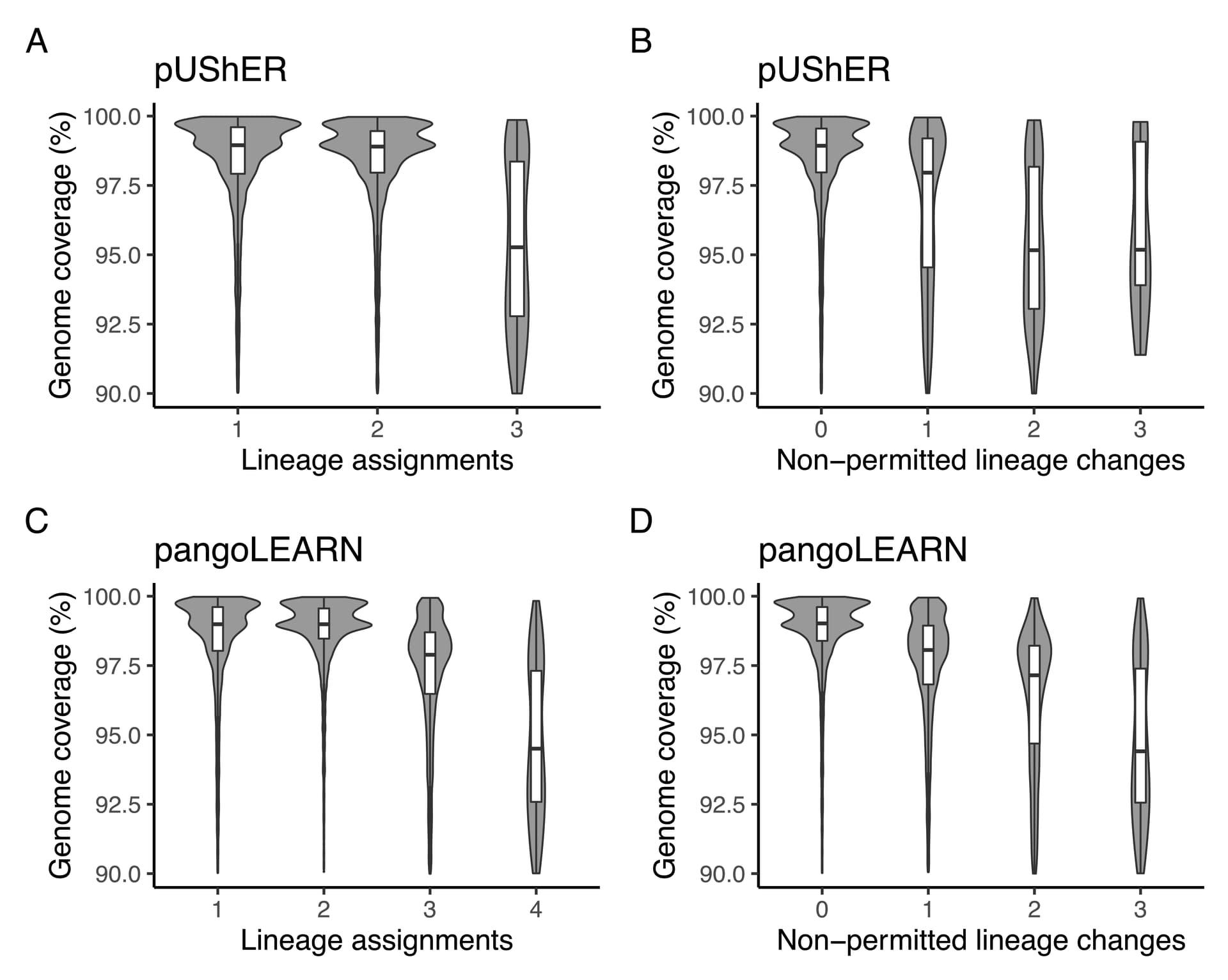
Figure 6. Violin plot of sample distribution based on genome coverage and pangoLEARN/pUShER lineage assignments. A: pUShER lineage assignment calls and reference genome coverage; B: pUShER lineage assignment non-permitted lineage changes and reference genome coverage; C: pangoLEARN lineage assignment calls and reference genome coverage; D: pangoLEARN lineage assignment non-permitted lineage changes and reference genome coverage.
Conclusion
For both of these recent datasets, UShER mode provided increased stability, reduced rate of non-permitted lineage reassignments, and there was a reduced impact of genome coverage on consistency of lineage calling. As a result, we recommend that the UShER option be the first choice when assigning lineages to newly and previously sequenced genomes. We stress, however, there are two important caveats to this recommendation. First, the lineage system is explicitly phylogenetic (1) and recently the Pango curation team has used a tree inferred with UShER to assign new lineages. We expect that this will have explicitly reduced noise in UShER lineage calls, both because the tree used to designate lineages is likely to be more stable over time than a phylogenetic tree generated using other methods, and also because only sequences which form coherent monophyletic clades on the UShER tree are designated. By contrast, there will always be a degree of noise within machine learning methods. However, if there are systematic biases associated with phylogenetic inference in UShER, then consistently inaccurate phylogenetic placements might create spurious lineage designations and assignments that appear to be consistent lineage calls. We consider this unlikely since UShER’s phylogenetic accuracy for SARS-CoV-2 phylogenetic inference is quite high (4,10).
Second, pangoLEARN is significantly faster than pUShER (0.03 vs 0.48 seconds per assignment on average) on a single thread, although pUShER is able to efficiently exploit parallelism to decrease this runtime. This difference is not significant when considering small numbers of sequences. Further, where cloud computing is available we estimate a complete reanalysis of 6 million genomes would cost approximately $43.44 on a typical amazon cloud instance (following Why is Usher mode not made primary mode for GISAID etc? · Issue #32 · cov-lineages/pangoLEARN · GitHub), if efficiently exploiting multicore architectures. This compute cost is relatively small compared to the total costs associated with producing a single genome sequence. However the difference in runtime does remain a limiting factor when pangolin is run on larger datasets using local or shared servers. We propose mitigating this in a future release of pangolin by introducing an UShER lineage assignment cache to rapidly assign lineages to sequences that have been seen before.
Acknowledgements
We would like to acknowledge the NYC DOHMH PHL staff; the CDPH COVIDNet Team and these CDPH members: Dr. Kathleen Jacobson, Dr. Carol Glaser, Dr. Mayuri Panditrao, Dr. Christina Morales, Dr. Nikki Baumrind, Elizabeth Baylis, Sabrina Gilliam; the University of California Office of the President, and COVIDNet WGS Lab Partners throughout California. COVID sequencing in NYC was supported (in part) by the Epidemiology and Laboratory Capacity (ELC) for Infectious Diseases Cooperative Agreement (Grant Number: ELC DETECT (6NU50CK000517-01-07) funded by the Centers for Disease Control and Prevention (CDC). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of CDC or the Department of Health and Human Services.. Genomic surveillance by COVIDNet was funded by Centers for Disease Control and Prevention, Epidemiology and Laboratory Capacity for Infectious Diseases, Cooperative Agreement Number 5 NU50CK000539.
The findings and conclusions in this article are those of the authors and do not necessarily represent the views or opinions of the California Department of Public Health of the California Health and Human Services Agency.
References
-
Rambaut, Andrew, et al. “A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology.” Nature Microbiology 5.11 (2020): 1403-1407.
-
Rambaut, Andrew, et al. “Addendum: A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology.” Nature Microbiology 6.3 (2021): 415-415.
-
O’Toole, Áine, et al. “Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool.” Virus Evolution 7.2 (2021): veab064.
-
Turakhia, Yatish, et al. “Ultrafast Sample placement on Existing tRees (UShER) enables real-time phylogenetics for the SARS-CoV-2 pandemic.” Nature Genetics 53.6 (2021): 809-816.
-
“pangoLEARN PANGOLIN 2.0: pangoLEARN description”. cov-lineages.org. Archived from the original on 4 November 2021. Retrieved 19 November 2021.
-
“Scorpio - serious constellations of reoccurring phylogenetically-independent origin”. GitHub - cov-lineages/scorpio: serious constellations of reoccurring phylogenetically-independent origin. Retrieved 12 December 2021.
-
Shu, Y., McCauley, J. “GISAID: from vision to reality” EuroSurveillance 22(13) (2017) doi:10.2807/1560-7917.ES.2017.22.13.30494 PMCID: PMC5388101
-
Katoh, Kazutaka, and Daron M. Standley. “MAFFT multiple sequence alignment software version 7: improvements in performance and usability.” Molecular Biology and Evolution 30.4 (2013): 772-780.
-
Minh, Bui Quang, et al. “IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era.” Molecular Biology and Evolution 37.5 (2020): 1530-1534.
-
Thorlow, Bryan, et al. “Online Phylogenetics using Parsimony Produces Slightly Better Trees and is Dramatically More Efficient for Large SARS-CoV-2 Phylogenies than de novo and Maximum-Likelihood Approaches.” bioRxiv DOI:https://doi.org/10.1101/2021.12.02.471004. Posted Dec. 3, 2021.