Due to length constraints on virological.org posts, we are splitting this report into two parts that should be read as a single report. This is Part 1, containing Background, Data, Methods, and Findings sections 1 through 5. See Part 2 for Findings sections 6 through 9, Concluding remarks, Acknowledgements, and References.
Michael Worobey, University of Arizona, Tucson, AZ 85705, USA
Karthik Gangavarapu, Department of Immunology and Microbiology, The Scripps Research Institute, La Jolla, CA, USA
Jonathan E. Pekar, Department of Medicine, University of California San Diego, La Jolla, CA, 92093, USA
Jeffrey B. Joy, Department of Medicine, University of British Columbia, Vancouver, BC, Canada
Louise Moncla, University of Pennsylvania, PA 19104, USA
Moritz U. G. Kraemer, Department of Biology & Pandemic Sciences Institute, University of Oxford, Oxford, UK
Gytis Dudas, Institute of Biotechnology, Life Sciences Center, Vilnius University, Vilnius, Lithuania
Daniel Goldhill, Department of Pathobiology and Population Sciences, Royal Veterinary College, London, UK.
Christopher Ruis, VPD Heart & Lung Research Institute, Department of Medicine, University of Cambridge, Cambridge, UK
Lorena Malpica Serrano, University of Arizona, Tucson, AZ 85705, USA
Xiang Ji, Department of Mathematics, Tulane University, New Orleans, LA 70118, USA
Kristian G. Andersen, Department of Immunology and Microbiology, The Scripps Research Institute, La Jolla, CA, USA
Joel O. Wertheim, Department of Medicine, University of California San Diego, La Jolla, CA, 92093, USA
Philippe Lemey, Department of Microbiology, Immunology and Transplantation, Rega Institute, KU Leuven, Leuven, Belgium
Marc A Suchard, Department of Biostatistics, University of California, Los Angeles, Los Angeles, CA 90095, USA
Angela L. Rasmussen, Vaccine and Infectious Disease Organization, University of Saskatchewan, Saskatoon, SK, Canada S7N 5E3
Meera Chand, UK Health Security Agency, London UK
Natalie Groves, UK Health Security Agency, London UK
Oliver G. Pybus, (1) Department of Pathobiology and Population Sciences, Royal Veterinary College, London, UK (2) Department of Biology & Pandemic Sciences Institute, University of Oxford, Oxford, UK
Thomas P. Peacock, The Pirbright Institute, Woking, UK, GU24 0NF; Department of Infectious Disease, Imperial College London, UK, W2 1PG
Andrew Rambaut, Institute of Ecology and Evolution, University of Edinburgh, Edinburgh, UK
Martha I. Nelson, National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20892
Background
During the winter of 2020-2021, a new genotype of highly-pathogenic (HP) H5N1 avian influenza A virus emerged in Europe, comprising a reassortant between the epizootic HP clade 2.3.4.4b H5N8 and local low pathogenicity wildfowl strains. This new genotype caused record levels of infections in farmed poultry throughout Europe and quickly traveled, via waterfowl flyways, into North America, Africa and East Asia (1–3). In following seasons, this panzootic genotype underwent further reassortment with local low pathogenicity avian strains from waterfowl or seabirds - in Europe, North America and beyond (2, 4, 5), generating a diverse range of genotypes. One of these North American reassortant genotypes then entered South America (6), and most recently, Antarctica (7).
This panzootic H5N1 lineage, and its descendent reassortant genotypes, has shown an unusually high propensity to infect mammals, particularly those of the order Carnivora. There have been a large number of reports of infections in domestic cats (8), wild and farmed foxes (9), farmed mink (10), and wild pinnipeds (6). Reported disease in these species is severe, often comprising neurological clinical signs, respiratory distress and death. During mammalian infections the panzootic genotypes very quickly gain mammalian adaptations in their polymerase genes, often within a single mammalian infection (11). Furthermore there is some evidence of limited or even sustained mammal-to-mammal transmission, particularly in mink farm outbreaks (9, 10), and among sea lions and fur seals in South America (6, 12).
However, the number and frequency of independent bird-to-mammal cross-species transmission remains highly uncertain due to the comparatively low level of field and genomic surveillance of the virus in wild birds. Furthermore, the mode of transmission in these events is unclear with one study showing that the strain affecting pinnipeds in South America was able to transmit between co-housed ferrets, but did not exhibit respiratory transmission (13).
Most recently a widespread outbreak of H5N1 has been detected in the USA in dairy cattle (14–16). Cattle have not previously been described as being an enzootic host for influenza A viruses, although there is some limited serological evidence they may become infected by human seasonal influenza (17) and are susceptible to H5N1 infection under experimental conditions, although they do not develop significant clinical disease (18). However, cattle do have an endemic influenza virus, Influenza D virus, which is a component of bovine respiratory disease complex (19). This current bovine H5N1 outbreak appears to be characterized by reduced milk production, severe mastitis, and mild respiratory disease, though the full range of its severity remains uncertain.
Data
In a separate post, we describe our efforts to assemble H5N1 consensus genomes from raw sequence read data submitted to SRA by USDA (239 samples, primarily cattle but also several additional species, deposited in BioProject PRJNA1102327).
We also collated metadata included in BioProject PRJNA1102327, including host species. Both our genome assemblies/consensus sequences, and a table of metadata associated with them, are available from our public GitHub repository (https://github.com/andersen-lab/avian-influenza.)
Notably, key metadata required for phylodynamic and phylogeographic inference related to the bovine H5N1 outbreak (date of sampling and geographic location more specific than country) were not included in BioProject PRJNA1102327. We accessed information in which US state samples were collected from and on which date for 152 out of the 239 samples (20), and included that in our table of metadata (21) and our analyses.
Additionally, we included 4 H5N1 viral genome sequences sampled in Texas in April (14) and deposited on BioProject PRJNA1092030 and three additional genomes sampled in Ohio in April shared with us by Andrew Bowman, Jim Lowe and Richard Webby. We combined this dataset with all complete North American 2.3.4.4b clade genome sequences available on GISAID from 2023-01-01 and later. These included genomes collected in Texas in dairy cattle, wild birds, domestic cats, and one human (A/Texas/37/2024(H5N1)) from 2024-03-10 onwards (14). GISAID acknowledgement table is provided (see below).
Resulting alignments are available at avian-influenza/alignments at master · andersen-lab/avian-influenza · GitHub. We are continuing to update this data set as new sequence reads appear on SRA, new genomic sequences appear on GenBank or GISAID, and new metadata becomes available.
Methods
Maximum likelihood phylogenetic inference. We excluded all genomes that had >10% Ns, resulting in 913 HA sequences, 872 PB1 sequences, 886 PB2 sequences, 917 MP sequences, 917 NP sequences, 911 NA sequences, 914 NS sequences, and 885 PA sequences. We used IQTREE2 (22) to perform phylogenetic inference on each genome segment, using a generalized time-reversible (GTR) nucleotide substitution model (23) with four gamma rate categories, an empirical nucleotide frequency distribution, and a minimum branch length of 1e-6 while collapsing polytomies. We midpoint rooted the resulting trees and visualized the trees using baltic (24).
We also inferred a maximum likelihood tree with bootstrap support values using 1,000 replicates, using the concatenated genome sequences of 243 H5N1 viruses that were collected from US dairy cattle or from other host species identified as closely related to the cattle viruses (i.e., positioned within the cattle clade or directly basal).
Natural selection. To explore potential changes in the selective regime since the MRCA of H5N1 in cattle, we analyzed the non-overlapping coding regions of each genome segment using RELAX across the maximum likelihood phylogenies (25). Non-avian and non-cattle virus sequences were excluded from the analysis, as was the stem lineage separating the avian and cattle viruses.
Bayesian phylogenetic inference. Molecular clock phylodynamic inference was conducted using a Bayesian approach in BEAST v1.10.5 (26) using the BEAGLE high-performance library (27). We subsetted the 913 HA genomes to those that included precise dates, and we excluded any non-bovine viruses that were nested within the cattle outbreak clade in the maximum likelihood HA tree, resulting in 715 genomes.
We enforced monophyly on the cattle viruses in order to employ a joint coalescent model with an exponential growth model on the cattle viruses and a constant population size on the remaining viruses. We used a GTR nucleotide substitution model with gamma-distributed site rate variation among sites with eight gamma rate categories and a strict molecular clock. We ran four Markov chain Monte Carlo (MCMC) chains for 50 million states each, sampling parameters and trees every 5,000 generations to continuous-parameter and tree log files.
We examined convergence using Tracer (28), removed 5 million states as burn-in, and then combined the parameter log files using LogCombiner. We ensured that the effective sample size of all scientifically relevant parameters was >200. Tree files were resampled at every 20,000 states and combined using LogCombiner. We created a maximum clade credibility tree using TreeAnnotator and visualized the tree using baltic.
We performed two sensitivity analyses: (i) including avian and non-human mammal viruses that nest inside the cattle clade and (ii) using solely the viruses sampled in cattle.
For (i), we augmented our initial dataset with 28 avian and non-human mammal viruses, resulting in 743 genomes in total, and used a single nonparametric skygrid coalescent prior, with the remaining parameterizations identical to the primary analysis. We used a GTR nucleotide substitution model with gamma-distributed site rate variation among sites with eight gamma rate categories and a strict molecular clock. We ran four MCMC chains for 50 million states each, sampling parameters and trees every 5,000 generations to continuous-parameter and tree log files. We examined convergence using Tracer, removed 5 million states as burn-in, and then combined the parameter log files using LogCombiner. We ensured that the effective sample size of all scientifically relevant parameters was >200.
For (ii), we subsetted the initial dataset to only include the viruses sampled in cattle (n=125) and used an exponential growth prior. We ran one MCMC for 10 million states, subsampling parameters and trees every 1,000 generations to continuous-parameter and tree log files. We examined convergence using Tracer and removed 1 million states as burn-in. The effective sample size of all parameters was >200.
Calculation and comparison of mutational spectra. To calculate reference spectra for mammals, wild birds and chickens, we initially reconstructed a phylogenetic tree on all available H5 HA sequences as of 10th August 2023, using iqtree with an HKY model of nucleotide substitution and four gamma classes. The goose-Guangdong lineage was extracted from this phylogenetic tree and its mutational spectrum calculated using MutTui (https://www.biorxiv.org/content/10.1101/2023.06.15.545111v1). To calculate spectra for each host group, we extracted mutations from this spectrum that occurred on tip phylogenetic branches sampled from the respective group. Significance of differences in mutation types between the spectra was examined using a permutation test where the difference in the mutation type proportion between the spectra was compared with that in 1000 random permutations of the mutations across spectra. The proportion of permutations with a difference at least as large as that in the real data was taken as a p-value, which was then corrected for multiple testing using the Benjamini Hochberg correction.
To calculate the cattle outbreak spectrum, we ran MutTui on each segment independently employing the alignment and ML phylogenetic tree described above. In each case, we calculated the cattle outbreak spectrum by labeling the phylogenetic tree into two groups: “outgroup” which consisted of all branches outside of the cattle outbreak and “cattle” which consists of all branches within the cattle outbreak. For the internal branches only spectrum, we split this cattle label into “internal” on internal branches within the clade and “tip” for tip branches within the clade, and retained mutations on the internal branch label. Segment spectra were combined using MutTui into a single outbreak spectrum.
We calculated the likelihood of generating the observed mutations under each of the mammal, wild bird and chicken spectra and compared mammal with wild bird or chicken through the likelihood ratio which we converted to a probability. To assess the ability of this method to robustly identify host where this is known, we random downsampled the mammal, wild bird and chicken spectra calculated above to the same number of mutations observed in the cattle outbreak (n = 487 in the full clade, n = 146 in the internal branches). 1000 downsampling were carried out and the probability of mammal vs wild bird or chicken calculated for each to generate a distribution.
Findings
Below, we list some noteworthy preliminary findings, based on analyses of the above sequence data and metadata.
1. A reassortment event within North American avian H5N1 2.3.4.4b viruses occurred shortly before the start of the cattle outbreak.
- The cattle sequences are all Genotype B3.13 [see GitHub - USDA-VS/GenoFLU: Influenza data pipeline to automate genotyping assignment for an explanation of genotypes].
- This genotype is a reassortant between the Eurasian panzootic H5N1 genotype and low pathogenicity North American genotypes first seen in late 2023. The PA, HA, NA and MP are derived from the European genotype and PB2, PB1, NP and NS derived from North American genotypes. See Figure 1 for a tanglegram of HA (which descends from the Eurasian panzootic H5N1 lineage) versus PB2 (which descends from a North American avian influenza genotype).
- This genotype is relatively rare in the USA but has been seen in birds as well as in wild mammals.
- Genotype B3.13 differs from the virus seen in a recent outbreak where H5N1 2.3.4.4b influenza A virus spilled over from poultry to goats. The outbreak in goats was unrelated to the current cattle outbreak.
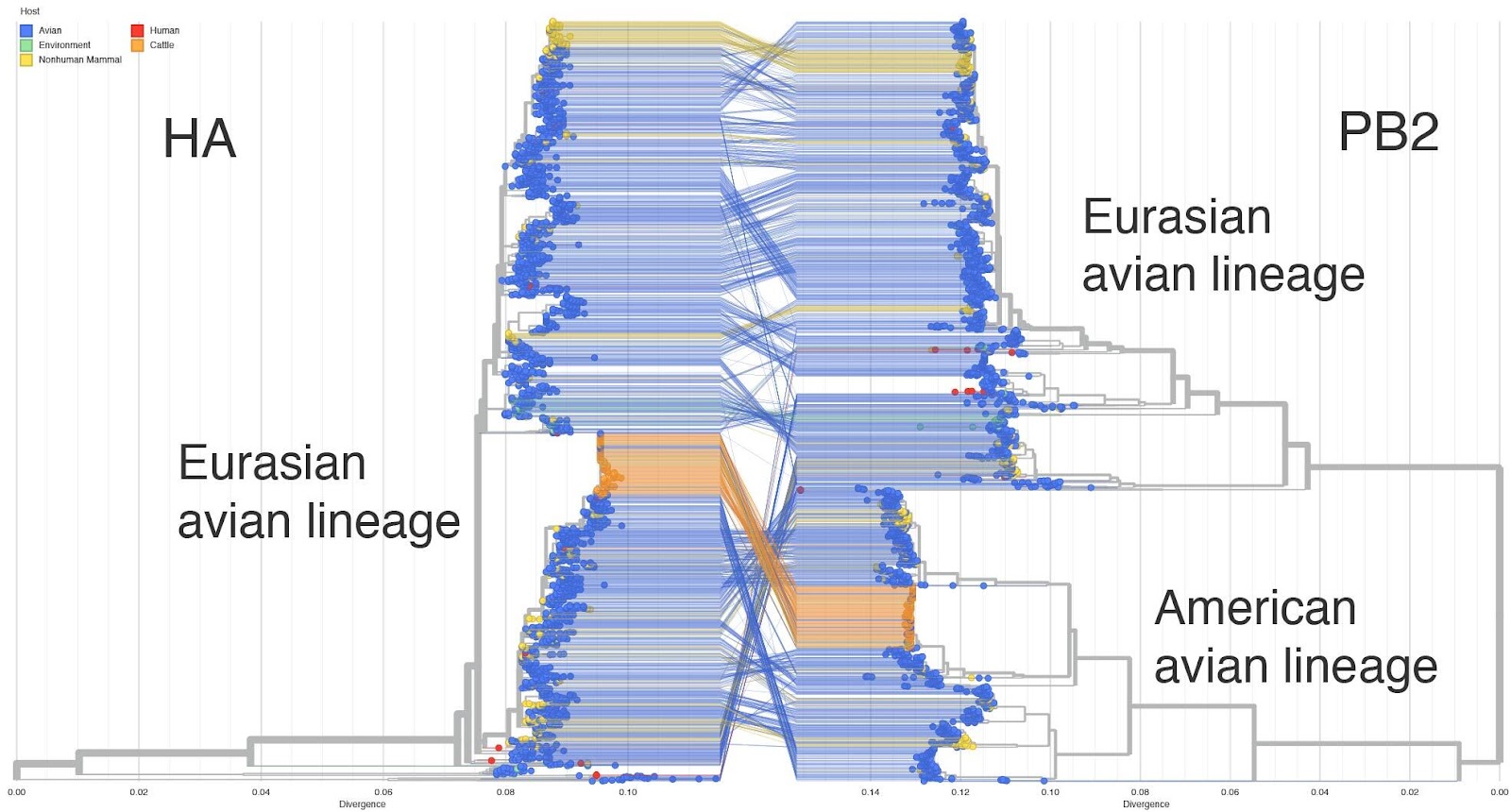
Figure 1. HA-PB2 Tanglegram - auspice.
2. The cattle outbreak likely had a single origin from the avian H5N1 reservoir.
- To determine whether the cattle outbreak arose from a single origin, we separately inferred maximum likelihood trees for each genome segment. We find that the viruses sampled from cattle form a monophyletic clade in each segment (Figures 2 & 3), consistent with a single introduction of H5N1 into cows and indicative of cattle-to-cattle spread.
- Had there been multiple jumps of H5N1 from birds into cattle, we would not expect to see monophyly of the viruses from cattle across the genome segments. Rather, we would expect to see a variety of constellations of gene segments because reassortment (mixing and matching among the 8 genome segments of the virus when individual hosts are infected by more than one strain) occurs frequently amongst avian influenza A viruses.
- See section 9, below, for additional evidence from mutational spectra.
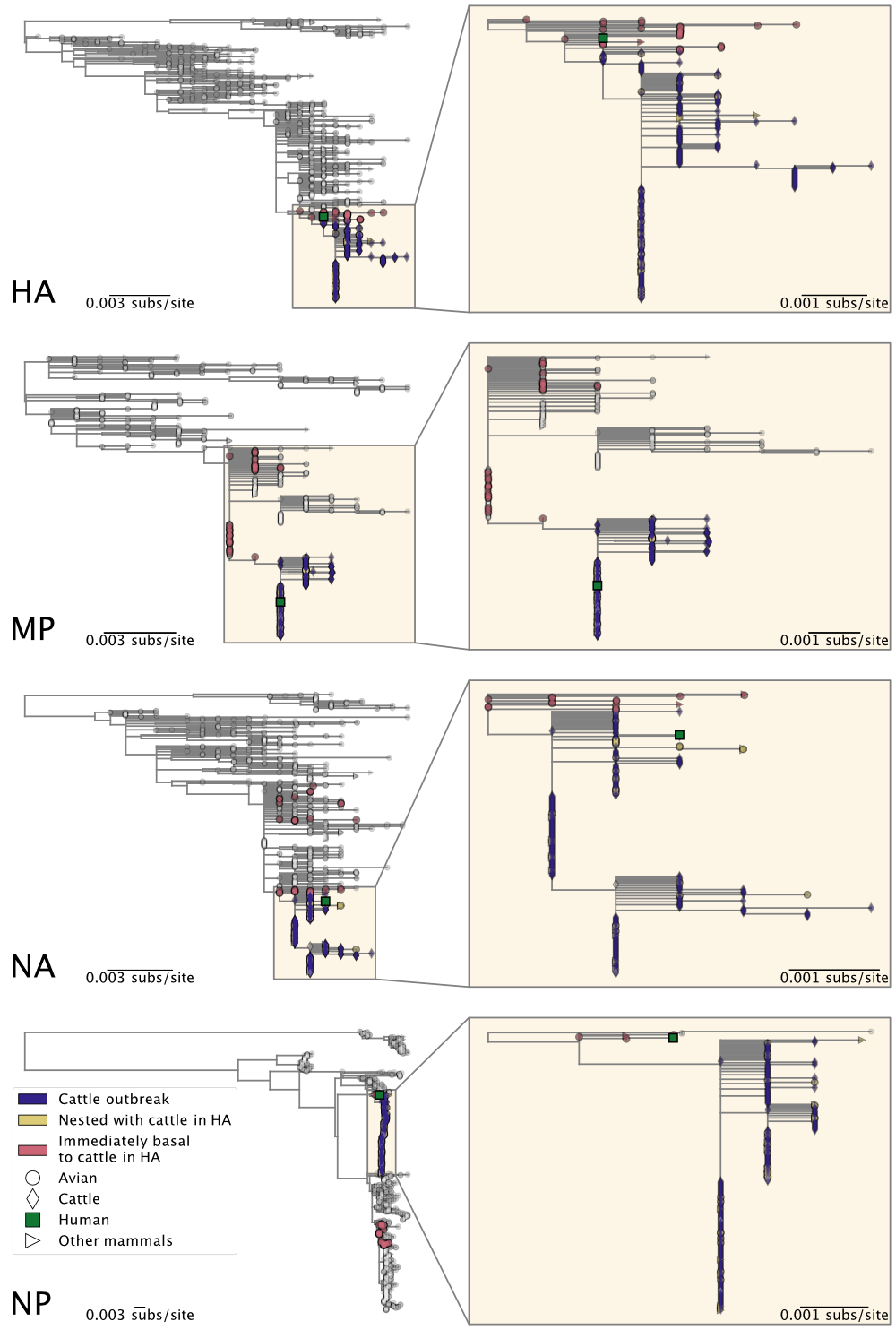
Figure 2. Maximum likelihood phylogenies of H5N1 2.3.4.4b genome segments HA, MP, NA, and NP. Inset on the right of each panel corresponds to the subtree, containing the cattle H5N1 sequences, highlighted in the larger tree on the left. See Figure 3 for all other genome segments.
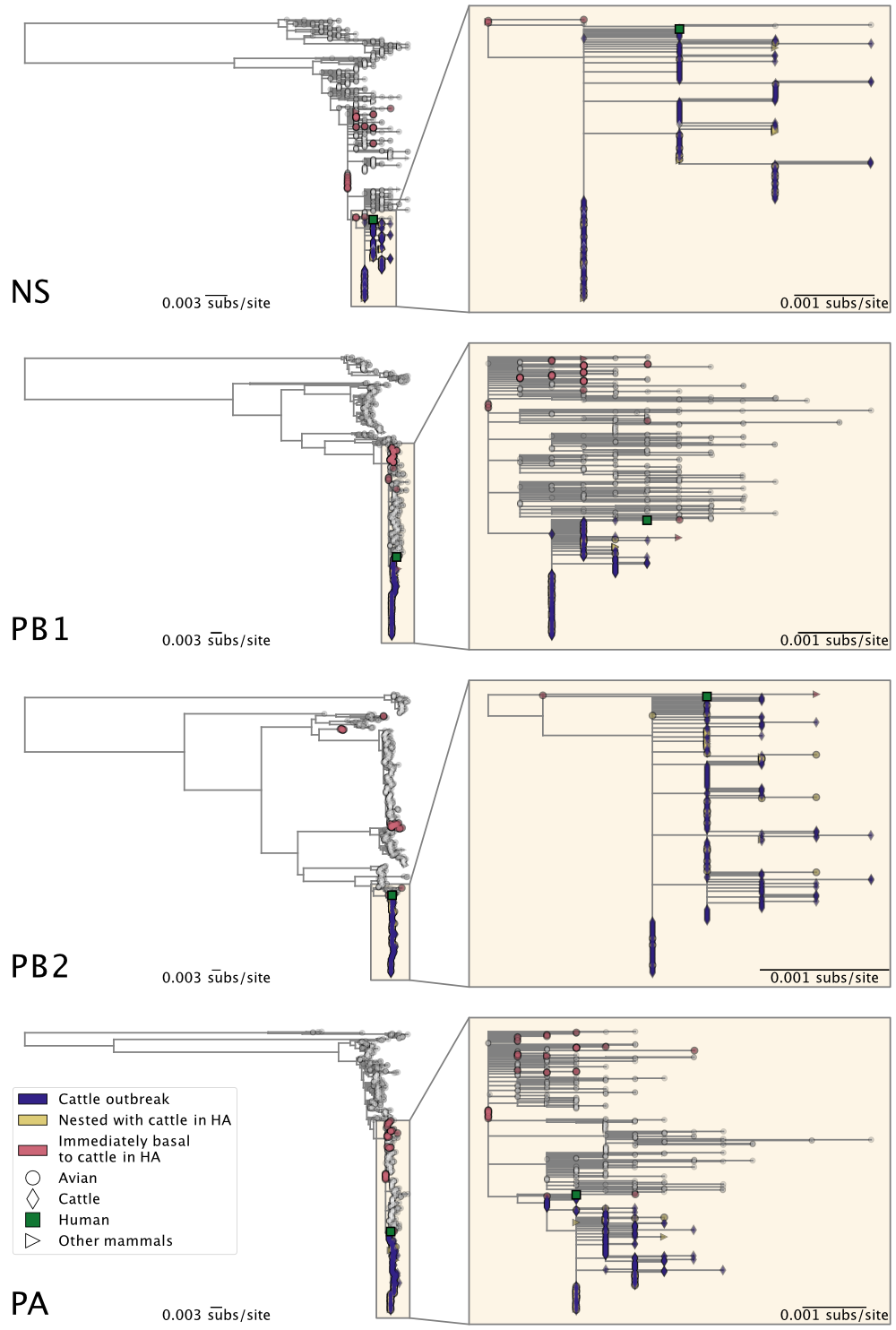
Figure 3. Maximum likelihood phylogenies of H5N1 2.3.4.4b genome segments NS, PB1, PB2, and PA. Inset on the right of each panel corresponds to the subtree, containing the cattle H5N1 sequences, highlighted in the larger tree on the left. See Figure 2 for all other genome segments.
3. The H5N1 outbreak in cattle likely went undetected and unidentified for an extended period and is now several months old.
- Based on the inference that this outbreak had a single origin in cattle and early molecular clock estimates, we hypothesized almost immediately after these sequence read data were released that the cattle H5N1 outbreak had already been going on for a long time and was very widespread in cattle (29). Accordingly, in a subsequent study across 38 states testing milk for influenza virus RNA, the FDA found that 1 in 5 samples were positive for H5N1 RNA(30).
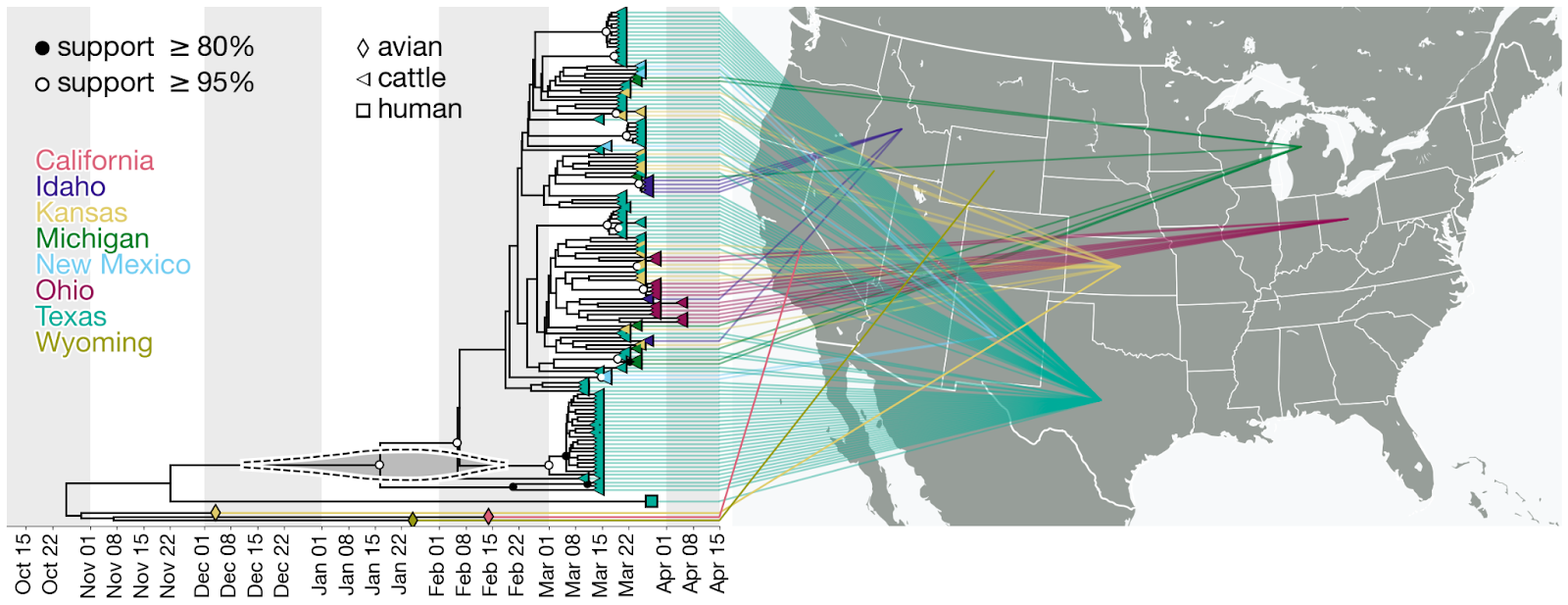
Figure 4. Time-calibrated phylogeny of H5N1 2.3.4.4b genome segment HA. Each genome is connected to the available geographic location in the United States. One genome sampled in the United States does not have state-level resolution and is colored the same as other genomes in North America. Violin centered on the most recent common ancestor (MRCA) of the viruses sampled in cattle represents the 95% highest posterior density (HPD) of the time of this most recent common ancestor (tMRCA).
- The jump from birds to cows must have occurred at some point along the branch leading to the common ancestor of the cattle viruses and sometime after the common ancestor of cattle viruses and their closest avian relative on the phylogenetic tree (Figure 5).
- To estimate the date of emergence of the virus in cattle, we reconstructed a time-resolved phylogeny (see Methods) using HA segments sampled from cattle with known collection dates and a background dataset of genomes from the 2.3.4.4b clade sampled in North America after 1 January 2023.
- After some initial analyses suggested relatively realistic modeling of a cattle H5N1 emerging from an avian reservoir might necessitate it, we employed two, separate, coalescent priors to better model the population dynamics within the cattle outbreak versus the background population of birds (and other mammals). Specifically, we used an exponential growth prior for the clade representing the cattle outbreak and a constant population size prior on the background sequences. Parameters under both priors were jointly inferred in the subsequent phylogenetic analysis. We estimated the median time to the most recent common ancestor (tMRCA) of the cattle clade as 18 January 2024 (95% HPD: [11 Dec 2023, 18 Feb 2023]). We estimated the tMRCA of the cattle clade and a related human virus as 22 November 2023 (95% HPD: [26 September 2023, 19 January 2024]). And we estimated the median tMRCA of the cattle clade and avian H5N1 as 13 November 2023 (95% HPD: [25 Sep 2023, 3 Jan 2024]).
- Hence, the jump from the avian reservoir into cattle likely happened between ~13 November 2023 and ~18 January 2024, meaning that the virus may have been circulating in cattle for up to 5 months before H5N1 was identified in them (Figures 5 & 6).
- On the other hand, given that the median tMRCA of the H5N1 cattle clade is mid-January, it is possible that the jump into cattle occurred only shortly before the earliest reported recognition of signs and symptoms consistent with H5N1 in late January 2024.
Figure 5. Schematic depicting the phylogenetic relationships between the HA segment of the viral genomes in different host species and when H5N1 likely spilled over into cattle.
- We infer an evolutionary rate of 6.73x10-3 (95% HPD: 5.79–7.72x10-3) substitutions/site/year. Our tMRCA is consistent with recently preprinted results, albeit slightly younger, which is possibly explained by our slightly higher inferred evolutionary rate (16). However, we note that our preliminary estimates were limited by the lack of known collection dates for over half of the raw samples deposited on SRA. As more metadata becomes available, we expect to more accurately infer the emergence of the cattle outbreak and further account for any potential variation in evolutionary rates across cattle and birds. If influenza virus evolves faster in cows than in birds, this might bias our estimate, leading to artifactually early estimates of introduction in cattle. Evolutionary rates of influenza A virus are known to differ between host species, and accounting for those differences can lead to more accurate reconstruction of evolutionary history(31)
- We performed sensitivity analyses using a nonparametric skygrid coalescent prior (32) for the same set of HA sequences and found that the tMRCA of the cattle clade was estimated to be 20 January 2024 (95% HPD: [24 December 2023, 13 February 2024]). However, in the same analysis we also infer a very similar tMRCA of the cattle clade and the human sequence sampled in Texas with a median of 17 January 2024 (95% HPD: [16 December 2023, 14 February 2024]). A separate inference on concatenated gene segments from cattle resulted in a late February 2024 tMRCA of the cattle clade (Table 1), along with an evolutionary rate of 1.61x10-2 (95% HPD: 1.25–2.05x10-2) subs/site/year, nearly 3-fold higher relative to the evolutionary rate inferred in our primary analysis. These later tMRCAs, a similar tMRCA of the cattle clade and the “human and cattle” clade, and the higher clock rate are not as plausible as our primary analysis and inconsistent with the circumstantial evidence surrounding the emergence of H5N1 in cattle. For instance, a tMRCA of the cattle H5N1 is at odds with reports that the disease was already being noticed by farmers and veterinarians in late January.
Table 1. Time of the most recent common ancestor (tMRCA) of cattle clade, tMRCA of cattle clade and human virus, and evolutionary rate for molecular clock phylogenetic analyses.
| Coalescent prior | Gene segment | Taxa included | tMRCA of cattle cladea | tMRCA of cattle clade and human virusa | Evolutionary rate (subs/site/yr)* |
|---|---|---|---|---|---|
| Joint constant and exponential | HA | All | 2024-01-18 [2023-12-11, 2024-02-18] | 2023-11-22 [2023-09-26, 2024-01-19] | 6.73e-3 [5.79–7.72e-3] |
| Skygrid | HA | All | 2024-01-20 [2023-12-24, 2024-02-13] | 2024-01-17 [2023-12-16, 2024-02-14] | 6.41e-3 [5.71–7.12e-3] |
| Exponential | All segments, concatenated | Cattle only | 2024-02-26 [2024-02-18, 2024-03-03] | N/A | 1.61e-2 [1.25–2.05e-2] |
*Median [95% HPD]
4. The cattle outbreak may have originated in Texas.
- The phylogenetic tree is consistent with an origin of the outbreak in Texas, where the first ill and first infected cattle were reported: the basal diversity on the tree is sampled in Texas (Figures 4 & 6). However, the vast majority of cattle samples are from Texas, with relatively few from other US states, potentially biasing this pattern. Given this potential sampling bias and our limited access to sampling location metadata, we did not conduct a formal phylogeographic analysis to infer movements of the virus across states.
- Within the cattle clade, we observe five well supported clades (with a posterior probability greater than 0.95) composed of genomes only from Texas (Figure 4). We also observe smaller clades with high posterior probability (>=0.95) composed of genomes sampled in other US states such as Kansas (two clades with two samples each), New Mexico (one clade with two samples), Idaho (one clade with four samples), and Ohio (one clade with four samples). Each of these clades could represent independent introductions of the virus into several states stemming from a common ancestor. We also observe a well supported clade with one sample from Texas and two from Michigan (Figure 4) indicating movement of the virus between these two states, but given the sampling bias, we refrain from drawing conclusions on the associated directionality.
- It cannot be ruled out that genotype B3.13 is especially prone to jumping into cattle and other mammals. Both the cattle H5N1 clade and a closely related virus sampled from an individual reportedly exposed on a dairy farm (see section 6, below) are from the same genotype and may have had separate origins from the primarily avian H5N1 2.3.4.4b reservoir.
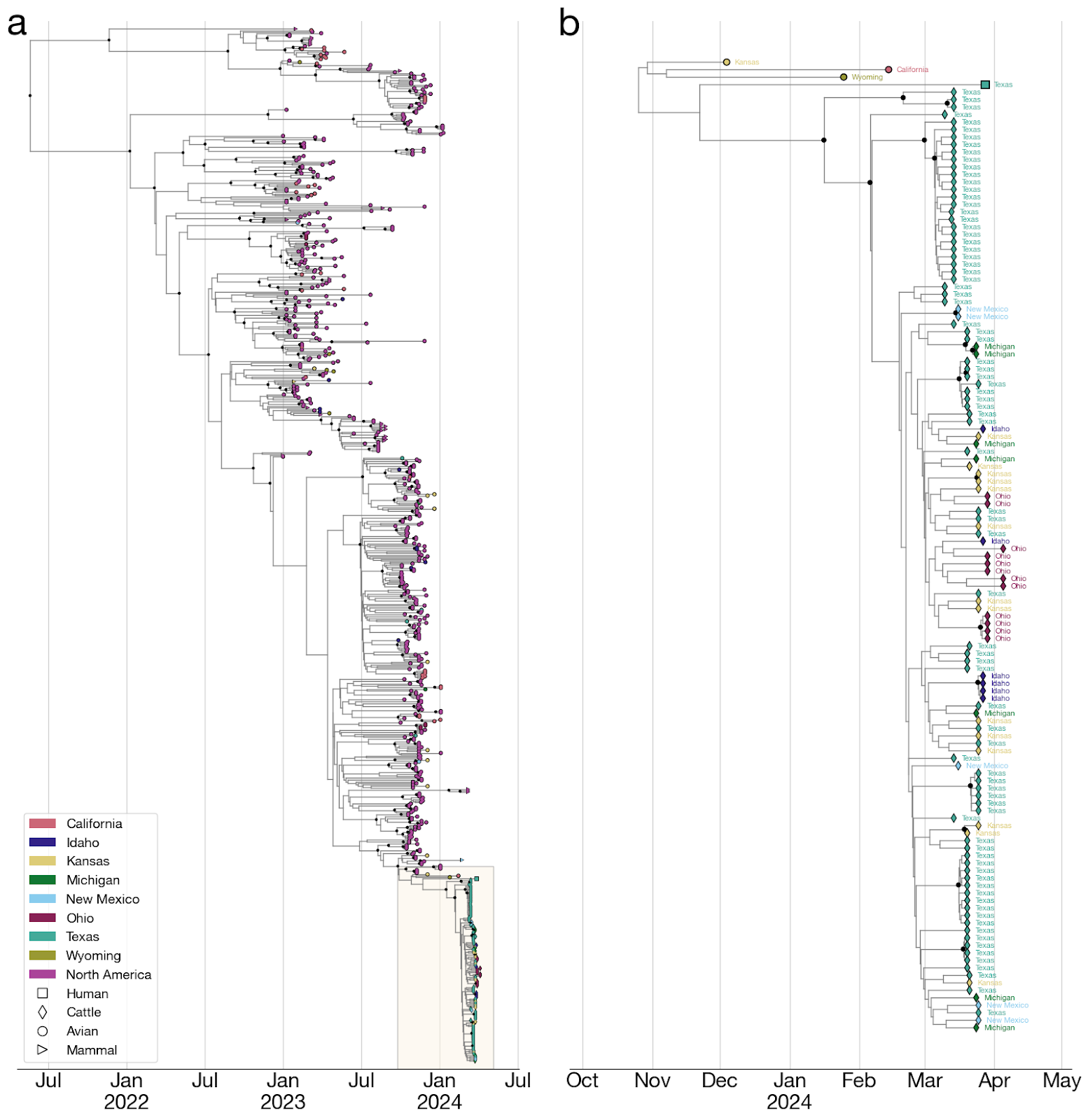
Figure 6. Time-calibrated phylogeny of H5N1 2.3.4.4b genome segment HA. a, Entire phylogeny. b, Subtree corresponding to highlighted inset in (a). This is the same phylogeny as in Figure 4, with taxon labels colored by state of origin (same as Figure 4).
5. Multiple putatively adaptive substitutions have arisen in the cattle H5N1 clade polymerase complex.
- For avian influenza viruses to productively infect mammals, they must overcome the poor activity of the avian virus RNA-dependent RNA polymerase in mammalian (including human) cells. Specifically they must adapt to use mammalian versions of a host protein called ANP32(33). The most well described ANP32-adapting mutation, PB2 E627K, is present in the human sequence but is absent in cattle sequences (though present as a minor variant in a single cow sequence from Kansas(16)). An alternative substitution, PB2 M631L, is present in cattle viruses but absent in the human virus(34). A few other putative ANP32 modulating mutations are present in the cattle viruses but are not fixed throughout the cattle cluster (Figure 7). PA K497R immediately precedes the main cluster of cattle sequences (clade C-E), while PA E613K (clade A) and PB2 E677G (clade B) are found in sister clades to those with PA K497R. PB2 M631L is very likely an adaptive mutation to mammalian ANP32 as it has been shown to interact with ANP32 and also to increase polymerase activity in human cells(35)(36). A recent influenza polymerase structure has shown PA K497 interacting with ANP32(36) and previous data has shown that this mutation can also increase polymerase activity(36). This pattern of substitutions is consistent with PB2 M631L arising early on during adaptation to cattle, with several different possible adaptive mutations in PA or PB2 likely arising during transmission among cattle (with PA K497R being in the majority of sequences).
- There is minimal evidence for a different selective regime acting within the cattle H5N1 virus clade compared with avian H5N1 viruses. Across the 8 segments, only PA exhibited a significantly different selective regime, with a modest increase in the intensity of selection (k=1.28; p<0.03). The selective regime across the other 7 segments did not significantly different between avian and cattle H5N1 viruses.

Figure 7. Amino acid changes mapped onto bovine H5N1 phylogeny, We inferred this maximum likelihood tree using 243 concatenated H5N1 genome sequences. H5 numbering is used for amino acid substitutions.