Phylogenetic analysis of nCoV-2019 genomes
23-Jan-2020
Andrew Rambaut, University of Edinburgh, Edinburgh UK
[email protected]
This post has been updated with new data here:
http://virological.org/t/phylodynamic-analysis-44-genomes-29-jan-2020/356/2
This is a brief report outlining a simple phylogenetic analysis of publicly shared genome sequences. It gives some preliminary findings for information purposes is not intended as an academic work. All the data used here is provided by the laboratories listed below through NCBI or GISAID.
Phylogenetic analysis
As of 23-Jan-2020, 24 full-length genomes are available on the GISAID platform. Two genomes are of insufficient quality to include in the analysis. 13 are from Wuhan City, Hubei, 4 from Shenzhen City, Guangdong, 2 from Zhuhai City, Guangdong, 2 from Zhejiang Province in China. An additional 2 genomes are we sampled in Thailand from individuals who had independently travelled from Wuhan. Acknowledgements and details of the genome sequences used in this analysis are given in Table 3 at the end of this document.
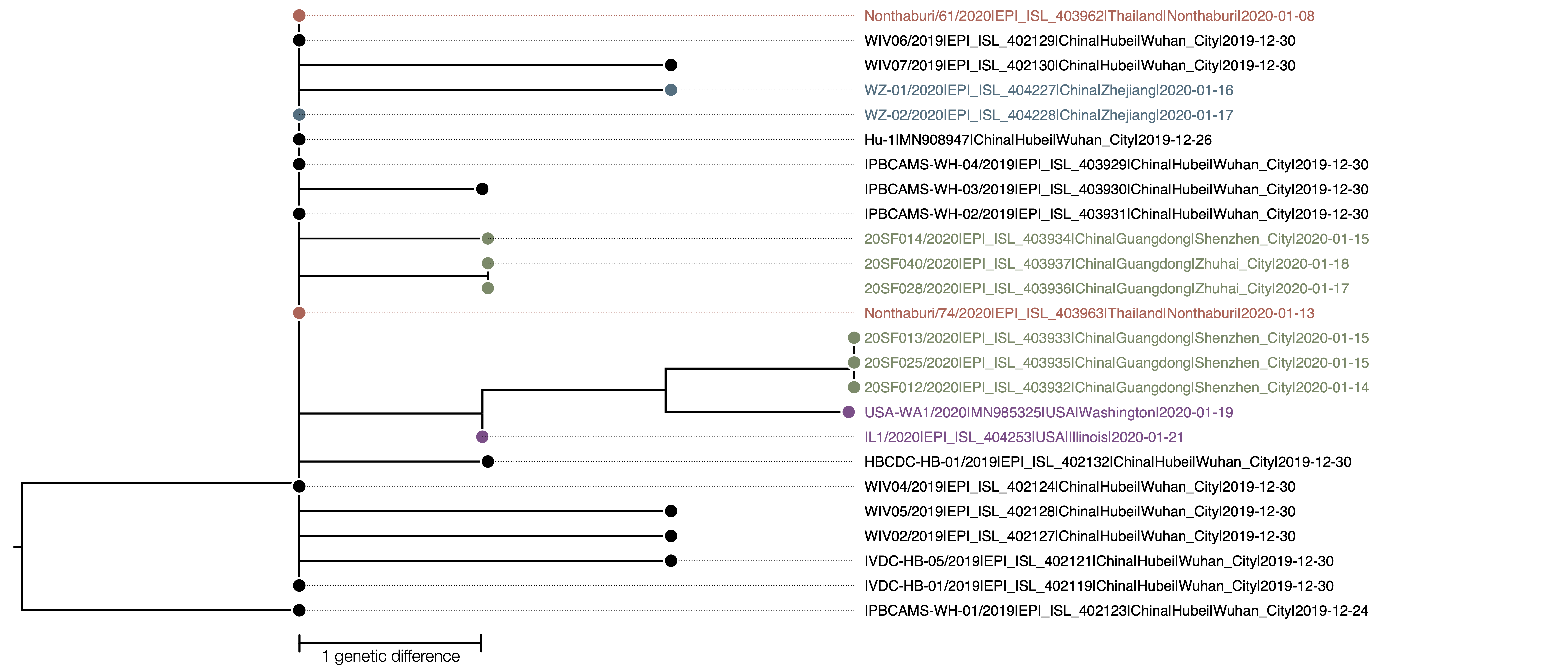
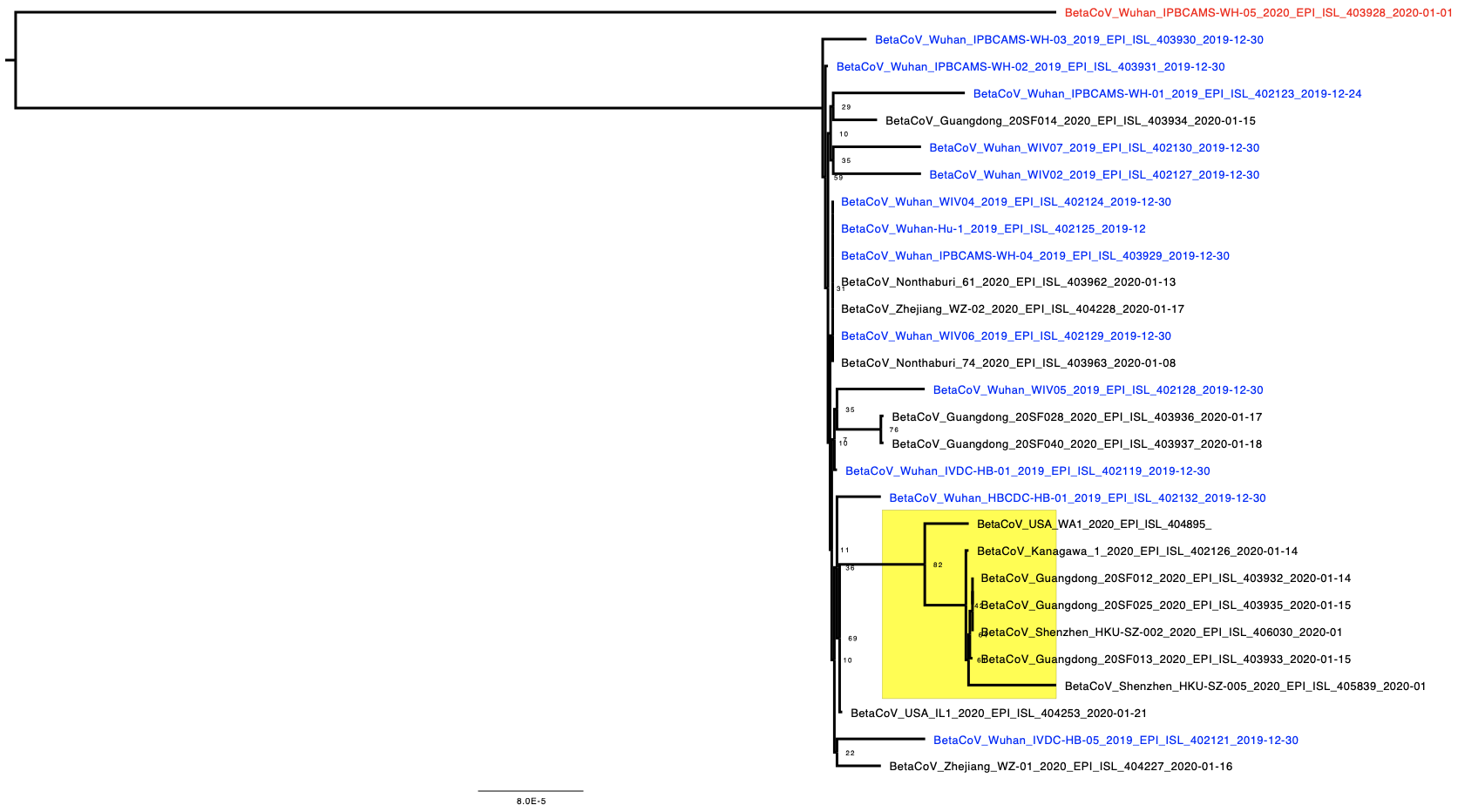
The phylogenetic tree of the currently available complete genomes is given in Figure 1. This shows that there is very limited genetic variation in the currently sampled viruses in Wuhan. This is indicative of a relatively recent common ancestor for all these viruses.

Figure 1 | Maximum likelihood tree of nCoV2019 genomes constructed using PhyML [1]. The tree is rooted using the oldest sequence but this is an arbitrary choice. The scale bar shows the length of branch that represents 1 nucleotide change in the genome.
The software package BEAST [2,3] was used to estimate the date of the most recent common ancestor (MRCA) of the currently available genomes. The MRCA represents the point where the ancestral virus of all the sampled cases was in the same host (whether this was a non-human animal or a human). At the moment, the rate of evolution for this virus is not-known so two likely extremes were used based on estimates made from related human coronaviruses (see Appendix for details).
The estimated dates for the most recent common ancestor (and the 95% credible interval) are compatible with the TMRCA at the beginning of December (Table 1). The earliest reported date of symptom onset for the initial cluster of pneumonia cases was 8th December 2019 [4].
| Assumed rate | Estimated date of MRCA | 95% interval |
|---|---|---|
| 1x10-3 | 29-Nov-2019 | 08-Nov-2019 – 16-Dec-2019 |
| 0.5x10-3 | 30-Oct-2019 | 18-Sep-2019 – 04-Dec-2019 |
Table 1 | The estimated date of the MRCA of the sampled nCoV-2019 genomes, given an assumed rates of 1x10-3 and 0.5x10-3 substitutions per site per year. Both of these rates give intervals that include the start of December.
Interpretation
The virus genomes sequenced thus far exhibit very little genetic variation which is indicative of a recent origin of the sampled and sequenced viruses.
The two genomes sampled in Thailand are genetically identical to 6 of the genomes sampled from Wuhan in late December. Given that there are no known epidemiological links with the Wuhan cluster it can be assumed that these two genomes representative of the viruses circulating at the time of exposure. This, in turn, suggests that the limit diversity present in the sampled and sequenced Wuhan cases is representative of the overall diversity of the outbreak at that time, supporting a recent origin of the human cases.
There is no evidence from these genome sequences alone that there has been additional zoonotic jumps from non-human animals after the initial Wuhan cluster in December but the number of sequences is very limited at present.
Caveats for the analysis
The number of genetic differences in the genomes is close to the error rate of the sequencing process. Some of the observed differences may be artefacts of this process in which case the genomes are more similar to each other.
The evolutionary rates used to estimate the TMRCA are supposed represent a plausible range based on previous estimates for other human coronaviruses.
The date estimates for the TMRCA is averaged over many plausible phylogenetic reconstructions of the genome data.
Appendix
To estimate the time of the most recent common ancestor (TMRCA) of the currently sampled viruses (including the ones from Thailand), I used the Bayesian phylogenetic software package, BEAST [3]. With the available data it is not possible to estimate the rate of evolution of the virus so I used two assumed values 1x10-3 substitutions per site per year (a reasonable expected rate of evolution for an acute RNA virus) and 0.5x10-3. These values approximately span the rate of rate estimates for other human coronaviruses shown in Table 2.
| Virus | Estimated rate x10-3 subst/site/year | Reference |
|---|---|---|
| SARS-CoV | 0.80 – 2.38 | Zhao et al. 2004 [2] |
| MERS-CoV | 0.63 [0.14 – 1·1] | Cotten et al. 2013 [3] |
| 1.12 [0.88 – 1.37] | Cotten et al. 2014 [4] | |
| 0.96 [0.83 − 1.09] | Dudas et al. 2018 [5] | |
| HCoV-OC43 | 0.43 [0.27 – 0.60] | Vijgen et al. 2005 [6] |
Table 2 | Evolutionary rate estimates of human coronaviruses
References
-
Guindon S, Gascuel O. A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Syst Biol. 2003;52: 696–704.
-
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29: 1969–1973.
-
Suchard MA, Lemey P, Baele G, Ayres DL, Drummond AJ, Rambaut A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018;4: vey016.
-
WHO | Novel Coronavirus – China. 2020 [cited 23 Jan 2020]. Available: http://www.who.int/csr/don/12-january-2020-novel-coronavirus-china/en/
-
Zhao Z, Li H, Wu X, Zhong Y, Zhang K, Zhang Y-P, et al. Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evol Biol. 2004;4: 21.
-
Cotten M, Watson SJ, Kellam P, Al-Rabeeah AA, Makhdoom HQ, Assiri A, et al. Transmission and evolution of the Middle East Respiratory Syndrome Coronavirus in Saudi Arabia: a descriptive genomic study. Lancet. 2013;382: 1993–2002.
-
Cotten M, Watson SJ, Zumla AI, Makhdoom HQ, Palser AL, Ong SH, et al. Spread, Circulation, and Evolution of the Middle East Respiratory Syndrome Coronavirus. MBio. 2014;5: e01062–13.
-
Dudas G, Carvalho LM, Rambaut A, Bedford T. MERS-CoV spillover at the camel-human interface. Elife. 2018;7. doi:(MERS-CoV spillover at the camel-human interface | eLife)
-
Vijgen L, Keyaerts E, Moës E, Thoelen I, Wollants E, Lemey P, et al. Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J Virol. 2005;79: 1595–1604.
Available genome data
| Accession | Strain | Location | Collection date | Lab |
|---|---|---|---|---|
| EPI_ISL_404227 | BetaCoV/Zhejiang/WZ-01/2020 | Zhejiang, China | 2020-01-16 | 1 |
| EPI_ISL_404228 | BetaCoV/Zhejiang/WZ-02/2020 | Zhejiang, China | 2020-01-17 | 1 |
| EPI_ISL_402132 | BetaCoV/Wuhan/HBCDC-HB-01/2019 | China/Hubei Province | 2019-12-30 | 2 |
| EPI_ISL_402127 | BetaCoV/Wuhan/WIV02/2019 | China / Hubei Province / Wuhan City | 2019-12-30 | 3 |
| EPI_ISL_402128 | BetaCoV/Wuhan/WIV05/2019 | China / Hubei Province / Wuhan City | 2019-12-30 | 3 |
| EPI_ISL_402129 | BetaCoV/Wuhan/WIV06/2019 | China / Hubei Province / Wuhan City | 2019-12-30 | 3 |
| EPI_ISL_402130 | BetaCoV/Wuhan/WIV07/2019 | China / Hubei Province / Wuhan City | 2019-12-30 | 3 |
| EPI_ISL_403963 | BetaCoV/Nonthaburi/74/2020 | Thailand/ Nonthaburi Province | 2020-01-13 | 4 |
| EPI_ISL_403962 | BetaCoV/Nonthaburi/61/2020 | Thailand/ Nonthaburi Province | 2020-01-08 | 4 |
| EPI_ISL_402120 | BetaCoV/Wuhan/IVDC-HB-04/2020 | China / Hubei Province / Wuhan City | 2020-01-01 | 5 |
| EPI_ISL_402119 | BetaCoV/Wuhan/IVDC-HB-01/2019 | China / Hubei Province / Wuhan City | 2019-12-30 | 5 |
| EPI_ISL_402121 | BetaCoV/Wuhan/IVDC-HB-05/2019 | China / Hubei Province / Wuhan City | 2019-12-30 | 5 |
| EPI_ISL_402124 | BetaCoV/Wuhan/WIV04/2019 | China / Hubei Province / Wuhan City | 2019-12-30 | 6 |
| EPI_ISL_402123 | BetaCoV/Wuhan/IPBCAMS-WH-01/2019 | China / Hubei Province / Wuhan City | 2019-12-24 | 7 |
| EPI_ISL_402125 | BetaCoV/Wuhan-Hu-1/2019 | China | 2019-12 | 8 |
| EPI_ISL_403931 | BetaCoV/Wuhan/IPBCAMS-WH-02/2019 | China / Hubei Province / Wuhan City | 2019-12-30 | 9 |
| EPI_ISL_403928 | BetaCoV/Wuhan/IPBCAMS-WH-05/2020 | China / Hubei Province / Wuhan City | 2020-01-01 | 9 |
| EPI_ISL_403930 | BetaCoV/Wuhan/IPBCAMS-WH-03/2019 | China / Hubei Province / Wuhan City | 2019-12-30 | 9 |
| EPI_ISL_403929 | BetaCoV/Wuhan/IPBCAMS-WH-04/2019 | China / Hubei Province / Wuhan City | 2019-12-30 | 9 |
| EPI_ISL_403937 | BetaCoV/Guangdong/20SF040/2020 | Guangdong, China | 2020-01-18 | 10 |
| EPI_ISL_403936 | BetaCoV/Guangdong/20SF028/2020 | Guangdong, China | 2020-01-17 | 10 |
| EPI_ISL_403935 | BetaCoV/Guangdong/20SF025/2020 | Guangdong, China | 2020-01-15 | 10 |
| EPI_ISL_403934 | BetaCoV/Guangdong/20SF014/2020 | Guangdong, China | 2020-01-15 | 10 |
| EPI_ISL_403933 | BetaCoV/Guangdong/20SF013/2020 | Guangdong, China | 2020-01-15 | 10 |
| EPI_ISL_403932 | BetaCoV/Guangdong/20SF012/2020 | Guangdong, China | 2020-01-14 | 10 |
[1] Department of Microbiology, Zhejiang Provincial Center for Disease Control and Prevention
[2] Hubei Provincial Center for Disease Control and Prevention
[3] Wuhan Institute of Virology, Chinese Academy of Sciences
[4] Department of Medical Sciences, Ministry of Public Health, Thailand & Thai Red Cross Emerging Infectious Diseases - Health Science Centre & Department of Disease Control, Ministry of Public Health, Thailand
[5] National Institute for Viral Disease Control and Prevention, China CDC
[6] Wuhan Institute of Virology, Chinese Academy of Sciences
[7] Institute of Pathogen Biology, Chinese Academy of Medical Sciences & Peking Union Medical College
[8] National Institute for Communicable Disease Control and Prevention (ICDC) Chinese Center for Disease Control and Prevention (China CDC)
[9] Institute of Pathogen Biology, Chinese Academy of Medical Sciences & Peking Union Medical College
[10] Department of Microbiology, Guangdong Provincial Center for Diseases Control and Prevention
Table 3 | nCoV2019 genome sequences used in this analysis, the GISAID accession numbers and submitting labs.