Something we wrote as part of a correction to the manuscript including S943P as a true variation:
As part of another study using a method to detect systematic sequencing errors (Freeman et al. 2020)(https://paperpile.com/c/iw1wsP/afZK), we are interrogating the quality of publically available sequencing data and this position was highlighted as suspect. We interrogated this position in the raw data of the Sheffield nanopore sequencing dataset. Although these two variants are not present in the final consensus sequence from any of the Sheffield isolates, the raw, untrimmed mapped sequencing reads show their presence in only one of the Artic amplicons covering the site (Figure1 A&B) in all samples analysed. We noticed that in fact this position is to the left of the 5’ primer of ampicon 81 in what we believe to be the nanopore adapter sequence. Comparison of the MN908947.3 reference and the adapter sequence reveals similarity around this position:
Nanopore adapter sequence:
CAGCACCTT
MN908947.3 reference sequence:
CAGCAAGTT
If this region was not trimmed before calling variants it is conceivable that because this region matches the reference and is not soft clipped by minimap2, the mismatch between the reference and the adapter sequence could be called as a variant.
In the Sheffield dataset validation set (Figure 1B), we see a C present at around 50% of called bases at both these positions in raw data but this region is trimmed by the Artic pipeline and is therefore not used to call variants and contribute to the final consensus sequence. It is also evident that although amplicon overlaps amplicon 81 in this region, there is no evidence for these variants in the data from amplicon 80.
In summary we believe this is an artefact that has arisen due to a combination of improper trimming of adapter and primer regions from raw sequencing reads before downstream analysis and the coincidental homology between the nanopore adapter sequence and the WuHan reference genome in this region.
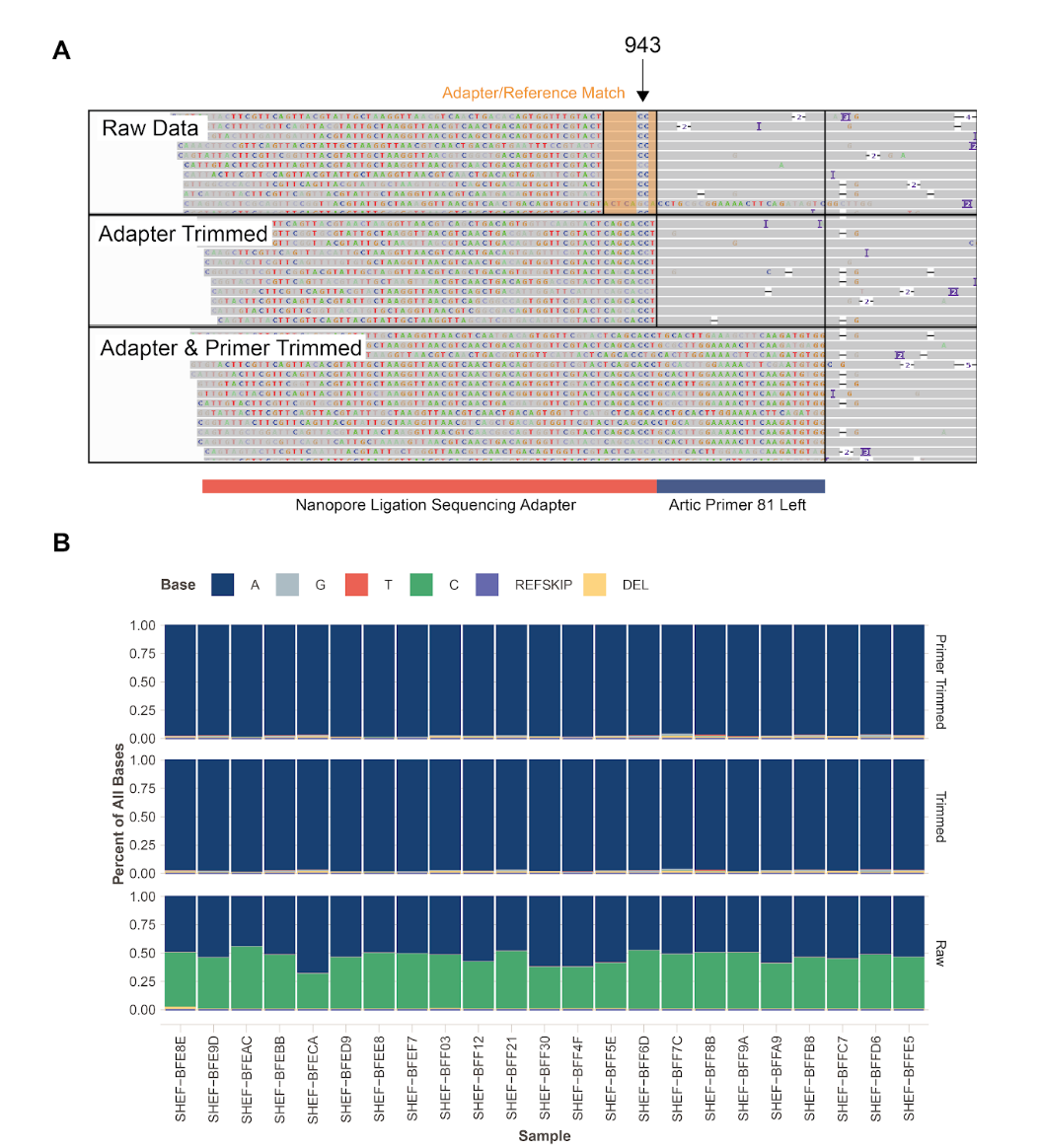
Figure 1 - Investigation of S943P
A. IGV plot showing bam files from nanopore sequencing data of amplicons produced by the Artic network protocol. Raw data from amplicon 81 contains adapter sequence which has high similarity to the reference genome, apart from the positions which lead to an erroneous S943P mutation call. This region is therefore included in variant calling if trimming is not carried out. Subsequent panels show that this region is soft clipped when trimming adapters and primers and is therefore not available for variant calling. B. Base frequencies at position 24389 in 23 samples from the Sheffield dataset show that C is present in half of the reads in the raw data, but is absent from trimmed and primer trimmed data.
Freeman, Timothy M., Genomics England Research Consortium, Dennis Wang, and Jason Harris. 2020. “Genomic Loci Susceptible to Systematic Sequencing Bias in Clinical Whole Genomes.” Genome Research 30 (3): 415–26.