Issues with SARS-CoV-2 sequencing data
Nicola De Maio1*, Conor Walker1, Rui Borges2, Lukas Weilguny1, Greg Slodkowicz3, Nick Goldman1
1European Molecular Biology Laboratory, European Bioinformatics Institute, Hinxton, Cambridgeshire, United Kingdom.
2Institut für Populationsgenetik, Vetmeduni Vienna, Veterinärplatz 1, Wien 1210, Austria.
3MRC Laboratory of Molecular Biology, Francis Crick Avenue, Cambridge Biomedical Campus, Cambridge CB2 0QH, United Kingdom.
*[email protected]
Summary
We investigate oddities in the SARS-CoV-2 genome sequences from GISAID. Many putative sequencing issues seem specific to genomic ends and to certain samples, and are easily filtered out. However, many mutations seem to arise many times along the phylogenetic tree (are highly homoplasic), and seem more likely the result of contamination, recurrent sequencing errors, or hypermutability, than selection or recombination. Some homoplasic substitutions seem laboratory-specific, suggesting that they might arise from specific combinations of sample preparation, sequencing technology, and consensus calling approaches.
To help other researchers in similar efforts or who rely on available SARS-CoV-2 genome sequences in downstream analyses, we summarize the steps that we have recognised so far useful for filtering and masking alignments of SARS-CoV-2 sequences. We also hope that this will spark a discussion regarding the best methods to identify and interpret such peculiar variants and samples, and we provide here a list of filters that so far we think reasonable.
First, we propose to mask alignment ends (as most of us already do), which are affected by low coverage and high rate of apparent sequencing/mapping errors. We mask positions 1–55 and 29804–29903 when aligned to reference MN908947.3, but other more or less stringent choices are possible.
Secondly, we propose masking sites that appear to be highly homoplasic and have no phylogenetic signal and/or low prevalence – these can be recurrent artefacts, or otherwise hypermutable low-fitness sites that might similarly cause phylogenetic noise. A current list of these is:
187, 1059, 2094, 3037, 3130, 6990, 8022, 10323, 10741, 11074, 13408, 14786, 19684, 20148, 21137, 24034, 24378, 25563, 26144, 26461, 26681, 28077, 28826, 28854, 29700.
We provide technical details of how these sites were identified below, however please note that all lists of sites outlined here are a work in progress, and might be affected by many choices made in the preliminary phylogenetic steps.
In addition, we suggest masking any homoplasic positions that are exclusive to a single sequencing lab or geographic location, regardless of phylogenetic signal. Here the phylogenetic signal might be caused by a common source of error (among other things). Our current list is:
4050, 13402.
We also recommend masking of positions that, despite having strong phylogenetic signal, are also strongly homoplasic. These may be caused by hypermutability at certain positions, although it is hard to rule out any possibility for now. Our current list is:
11083, 15324, 21575.
Finally, as other groups have already suggested (see e.g. [12]), we recommend filtering out sequences that: have too few resolved characters (our somewhat arbitrary threshold is about 29,400 reference bases), are too diverged (as can be tested using TreeTime), have unusual locally high divergence (as can be tested using ClonalFramML), have missing/incomplete sampling date information, or that are distant from any other sequence in the dataset (we use a custom script to remove all sequences that are at least three substitutions away from any other sequence). We don’t provide a current list as this is quite long and varies as the number of publicly shared SARS-CoV-2 genome sequences increases.
Detailed analyses
GISAID alignment preparation
We downloaded 5894 SARS-CoV-2 consensus genomes from GISAID [1] on 11th April 2020, and removed animal samples (bat and pangolin). We also removed sequences with less than 29,400 informative nucleotides. The remaining sequences were aligned with MAFFT v7.453 [2] (options --auto --keeplength --addfragments ) to reference genome MN908947.3, which is 29,903bp long. We then masked genome positions covered by less than 90% of the sequences in the alignment (genomic ends 1–55 and 29804–29903) as these low-coverage positions are seemingly prone to putative alignment and/or calling errors (see e.g. Figure 1). So far these filters are quite obvious and have been used, in some version, by other studies so far.
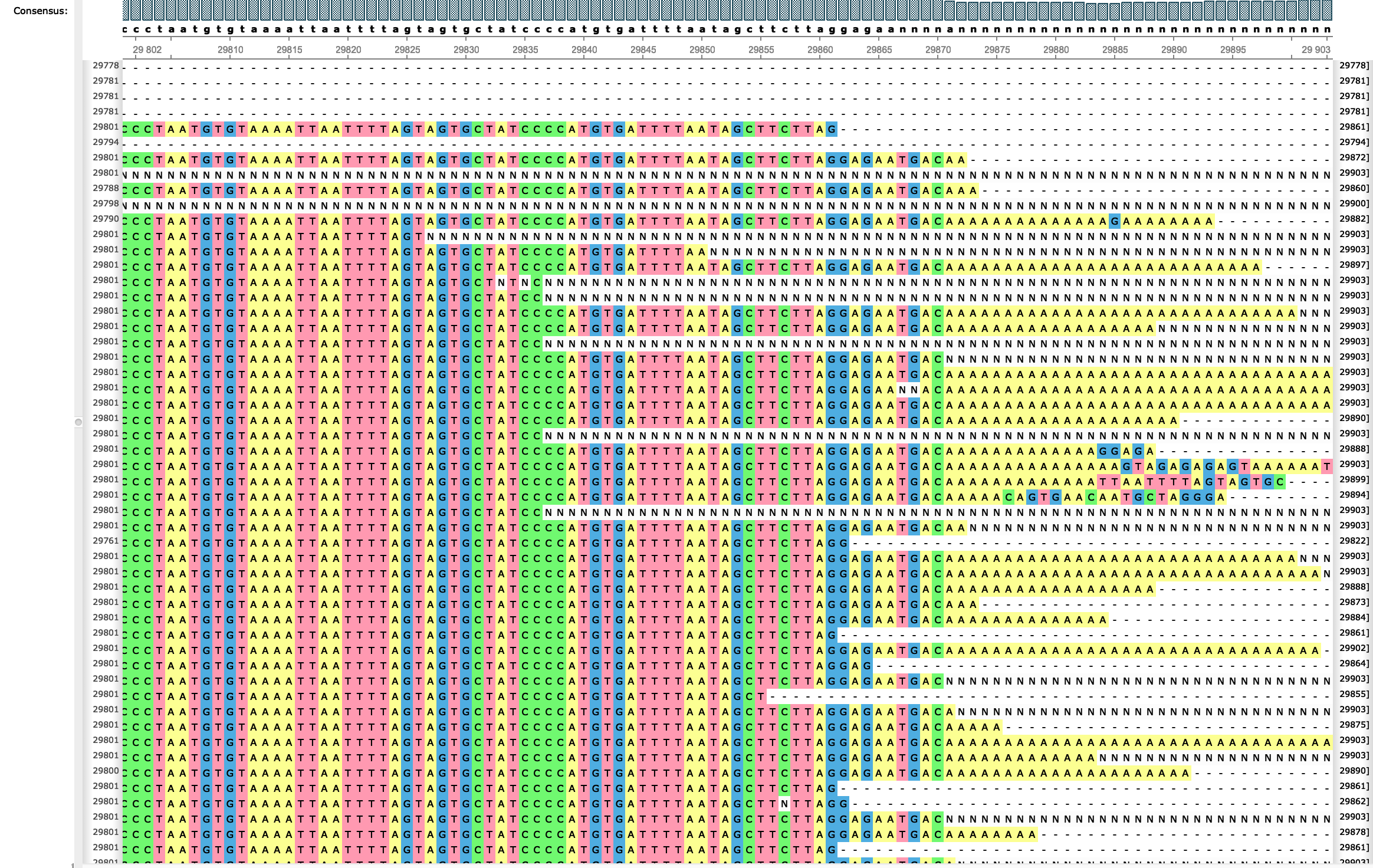
Figure 1: right end of a MAFFT alignment showing seemingly problematic alignments at the end of the genome.
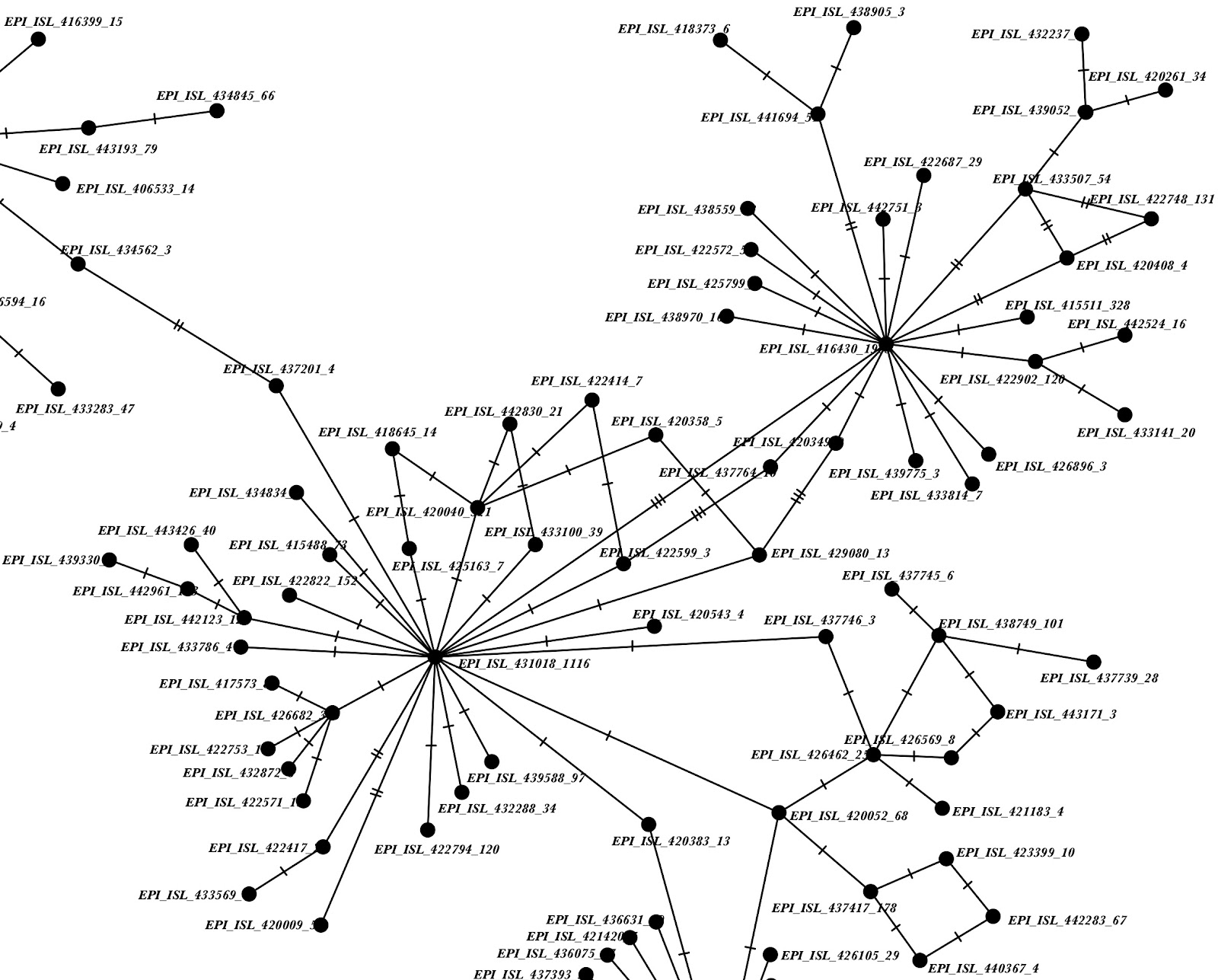
We then ran IQTree v1.6.12 [3] (options -st DNA -m HKY) on the resulting alignment. A few samples have extremely long terminal branches (Figure 2), suggesting either evolutionary events leading to many substitutions (e.g. recombination events or large mutation events), or sequencing/calling artefacts in specific samples.
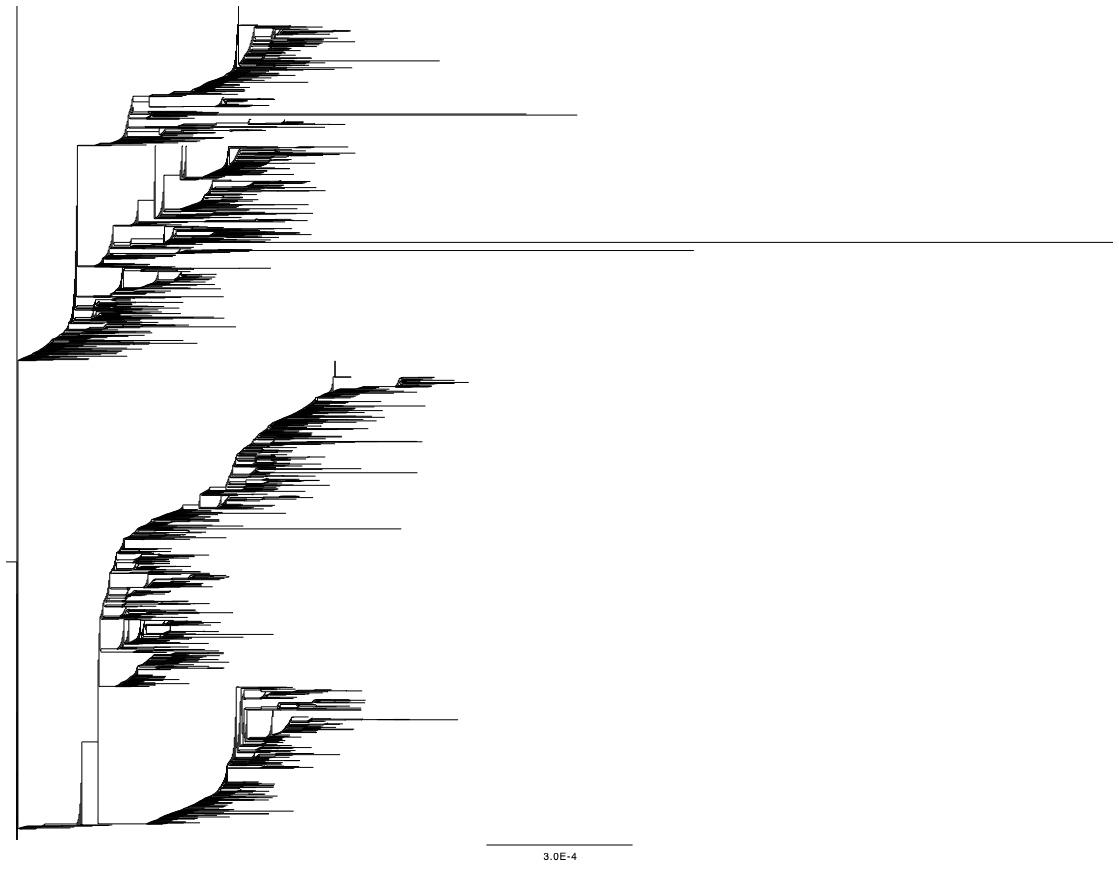
Figure 2: IQTree phylogeny showing some very long terminal branches.
Investigating recombinations and clusters of mutations/artefacts
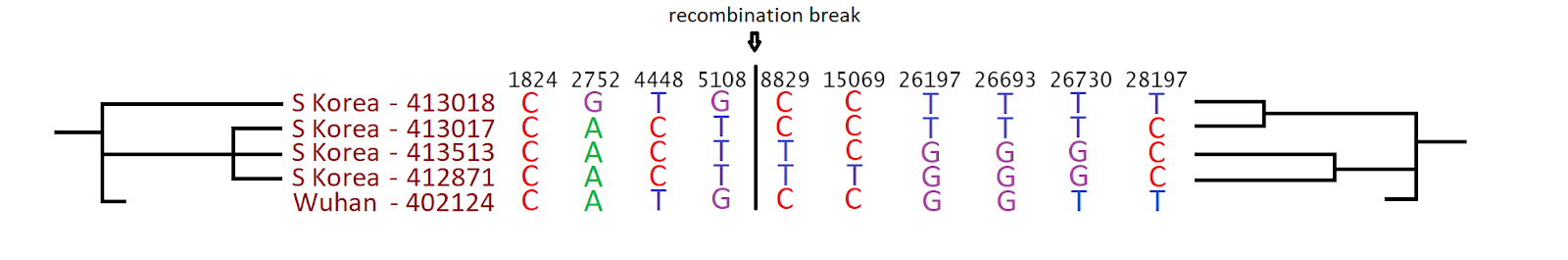
To further investigate, and to look into possible recombination events, homoplasies (mutation events seemingly happening multiple times along the phylogeny) and mutational clusters, we ran ClonalFrameML v1.12 [4]. This software found 30 putative recombination events happening at terminal branches, consistent with the long terminal branches in Figure 2. These putative events appear as clusters of substitutions in individual samples and genomic positions (e.g. Figure 3). Given that these mutations are not observed in any other samples, they could represent artefacts in the corresponding sequence. We remove the samples presenting these mutational clusters from further analyses; this filtering strongly overlaps with the filtering of samples showing inconsistent temporal signal in TreeTime [5] (see below).
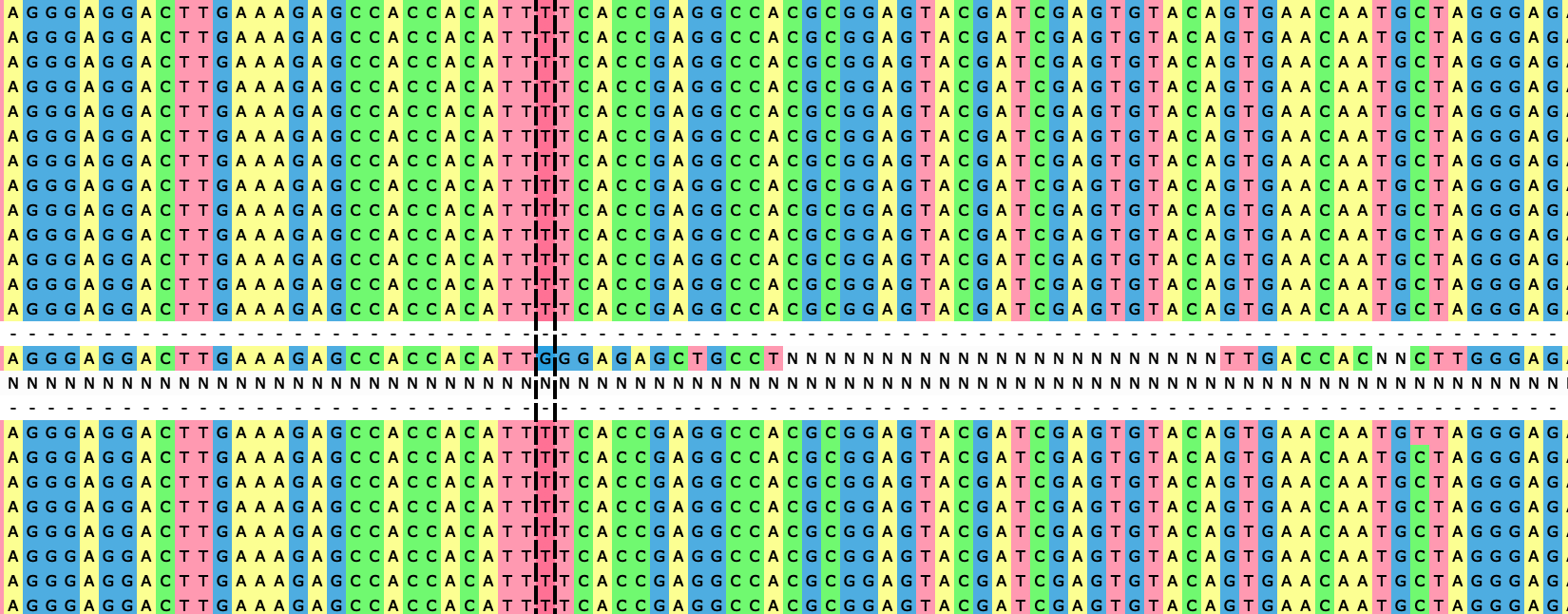
Figure 3: example of a mutation cluster in a terminal branch.
ClonalFrameML also suggests 3 putative recombination events (position 6971–6979, 13686–13693, and 28881–28883) happening on internal branches, however, these are all very short, and contain mutations not found on other branches of the tree (they are not homoplasies), and therefore these are more likely clusters of substitutions caused by individual multi-nucleotide mutations. We masked these mutational clusters from the alignment, as these events can bias classical phylogenetic and evolutionary analyses. The most striking of these putative multi-nucleotide mutations is 28881–28883 (Figure 4), replacing GGG with AAC. This is the only one of these mutations reaching high frequency in the population (776 sequences, or 16.5% of the samples, while 6971–6979 and 13686–13693 only appear in 2 samples each). The three substitutions at 28881–28883 seem to only appear in complete linkage, with the exception of G28881A that also appears in another two samples. (However, these samples were later filtered from the dataset in following steps.) Another interesting aspect is that the 28881–28883 mutation also seems to appear in other non-phylogenetically related samples as within-host polymorphism (Figure 4). This hints at a considerable proportion of cases being either mixed infections (patients infected with multiple strains of SARS-CoV-2) or maybe in some cases contaminated at low frequency.
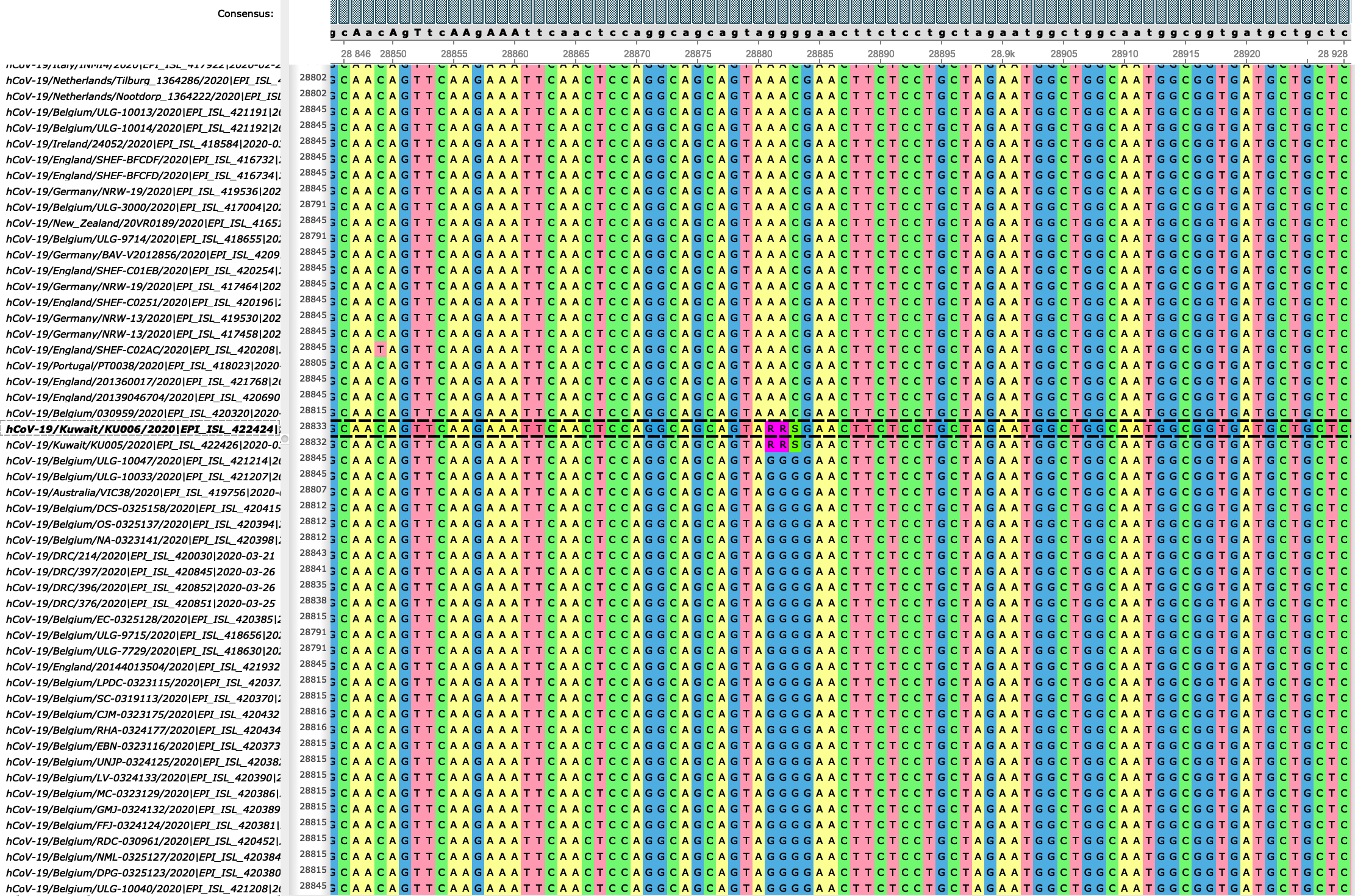
Figure 4: alignment extract around positions 28881-28883 showing the 3-nucleotide substitution. The reference GGG haplotype or the alternative one AAC are present in most samples. In some samples, like the highlighted one, however, show “RRS” at these positions: these are the ambiguity codes for the reference and alternative alleles combined, suggesting that both alleles were present in the same sample.
In addition to these clusters of mutations, ClonalFrameML also reveals homoplasies, mutations apparently appearing multiple times along the phylogeny. When multiple homoplasies happen on the same branch, they are the hallmark of recombination. However, we only observe isolated homoplasies (Figure 5), suggesting either recurring mutations, or recurrent sequencing/calling artefacts (see e.g. [6]), rather than very short recombination events at given hotspots, as suggested by some previous studies (see e.g. [7-8]). We investigate these homoplasies further in the next section.
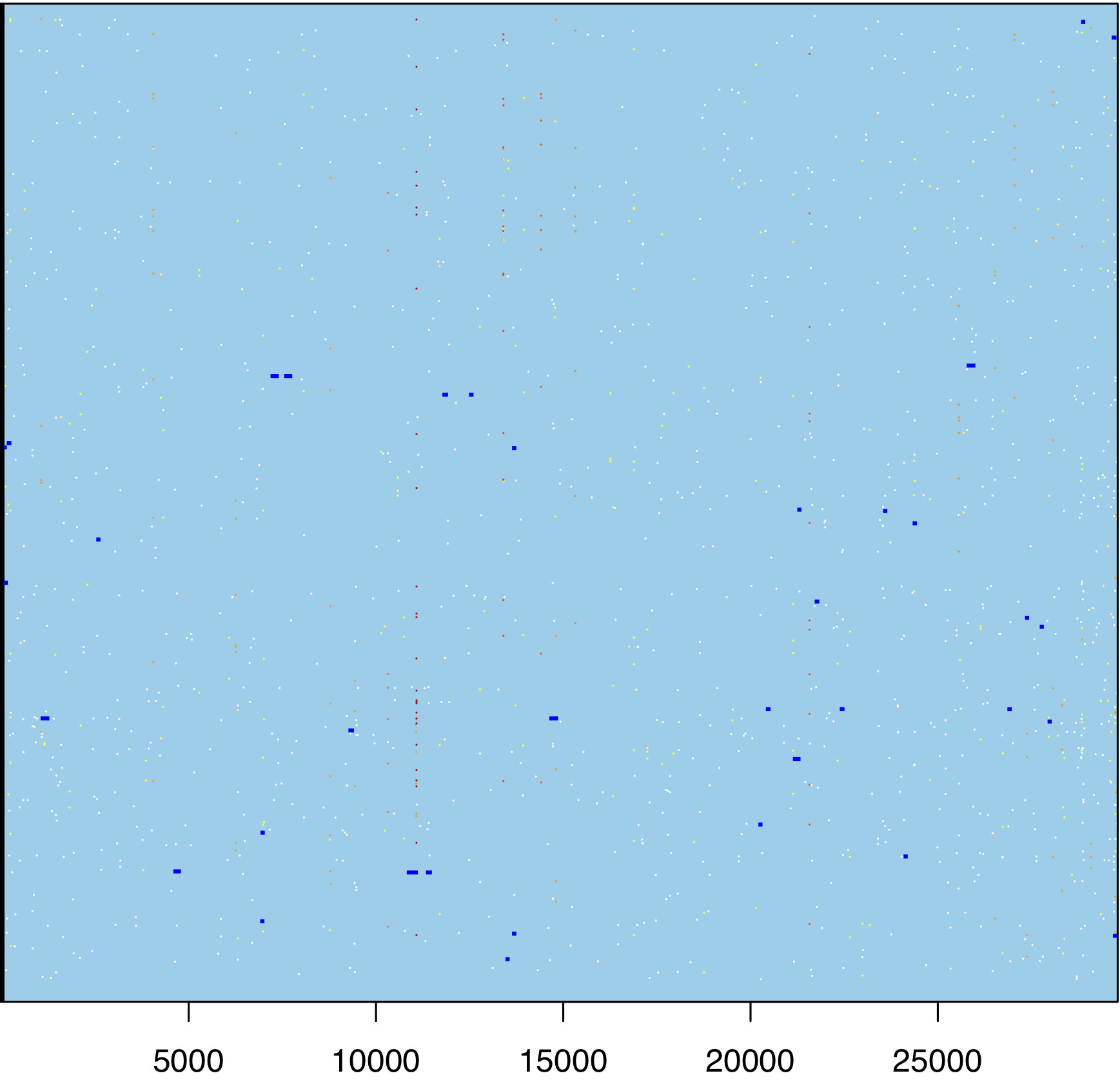
Figure 5: ClonalFrameML representation of putative recombinations (dark blue segments, later found to be clusters of mutations masked from downstream analyses), normal substitutions (white dots) and homoplasies (yellow to red dots, red being stronger homoplasies). Zoom in to see the individual dots. On the X axis are genome positions; the Y axis shows different tree branches (the phylogeny is not shown).
We repeated the ClonalFrameML analysis after the removing the clusters of substitutions found above, and specifying strong priors so to favour the detection of large recombination events at low levels of divergence. No further recombination events were inferred.
Spectrum of mutations
We find a total 2775 SNPs, of which 1085 are non-singletons (i.e. appearing in at least 2 samples), and of which 45 have at least 1% frequency. The mutational spectrum of new mutations seems enriched in C→T and G→T mutations (Figure 6), and while T is the most frequent nucleotide in the genome, its frequency seems to be further increasing, at least before population-level selection is accounted for. Also, the substitution process seems strongly non-reversible and not at equilibrium.
For a more in depth analysis of how read processing pipelines affect the called mutations, see Rayko and Komissarov [13].
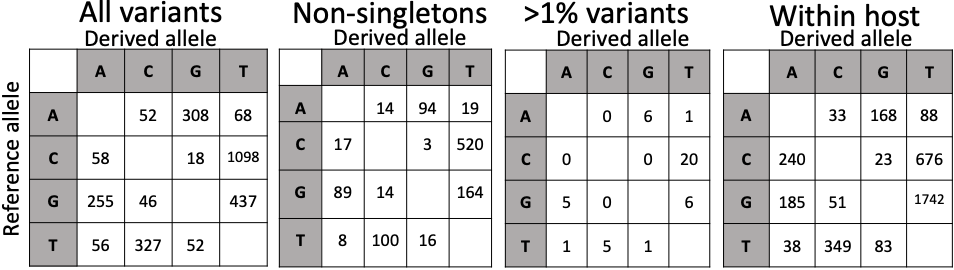
Figure 6: numbers of variants of each type classified based on the ancestral allele (row) and the derived allele (column). Here the reference allele is assumed to be ancestral for simplicity, but alternative roots do not change the overall patterns. The last table on the right is from within-host read data (see below).
As additional filtering steps, we removed duplicate samples, samples with incomplete date information, and highly diverged samples as filtered by NextStrain [9] (https://github.com/nextstrain/ncov/blob/master/config/exclude.txt), but not those excluded due to uncertain geographic origin. We also used TreeTime v0.7.5 to test and remove sequences with a genetic distance from the root that is significantly higher than expected based on their sampling time.
Sequencing technology, sequencing lab, and homoplasies
As mentioned before, we noticed many homoplasies in this dataset, more than expected by chance (see Figure 7, e.g. 33 sites with 4 mutations compared to expected 0.24 of them), and more homoplasic than expected by chance (e.g. 5 sites hit by more than 9 mutations, while no such site should have been observed). Baseline null distributions were calculated using the Poisson null in TreeTime, which however ignores some complications such as the effects of selection and possible hypermutable positions. Some of these mutations, particularly G11083T, seem geographically ubiquitous, and appear in consensus sequences generated by any technology. Position 11083 seems to have mutated 28 times, both from G to T (21 times) and back from T to G (7 times).
Other mutations are Illumina or nanopore-specific, and some seem specific to both nanopore and to some countries (for example T13402G, A4050C, T13408C, T8022G, C3130T, T28785G).
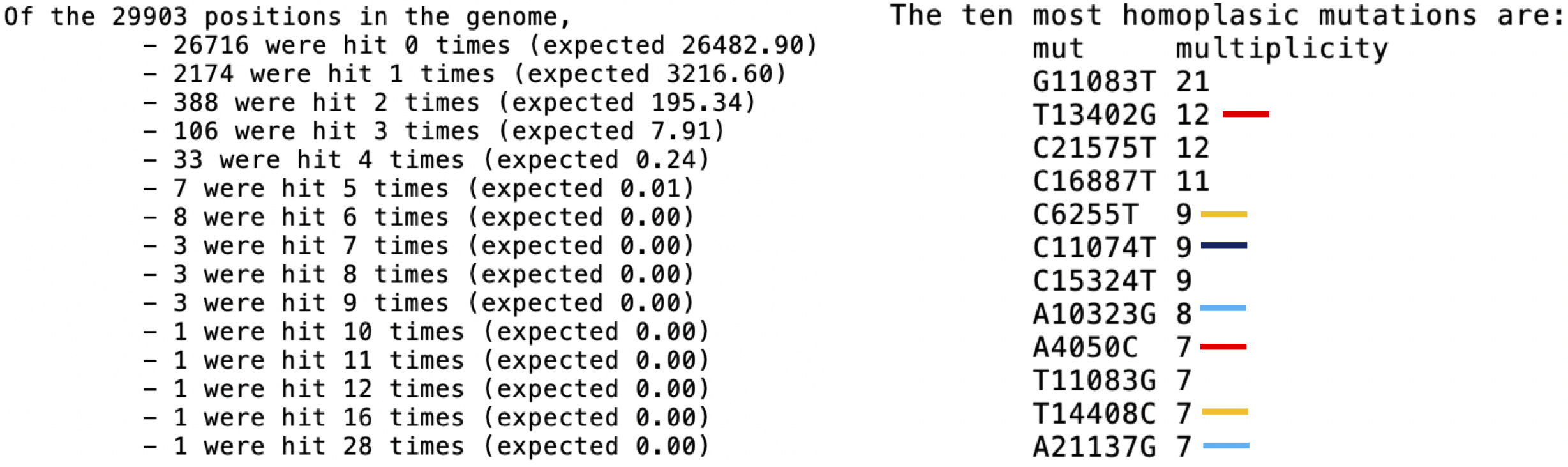
Figure 7: TreeTime homoplasy results. On the right, only the most homoplasic mutations are shown. Red lines: only present in ONT; orange: only homoplasic in ONT; dark blue: only present in Illumina; light blue: only homoplasic in Illumina.
To investigate this pattern further, we split our dataset into 3 groups: (A) Illumina sequences (n=2253), (B) nanopore sequences not sequenced at KU Leuven (n=550), and (C) nanopore sequences sequenced at KU Leuven (n=186). We only included sequences from labs that submitted large numbers of sequences consistently sequenced with the same technology. C nanopore sequences were from USA, England, Belgium, Netherlands, Germany and Canada. Illumina sequences were from USA, China, Australia, Japan, Singapore, France, DRC, Luxembourg, Portugal, Wales, Canada and Iceland.
We re-analysed these 3 datasets individually using TreeTime. The most predominant homoplasies were common across technology and country, including G11083T, C16887T, C21575T and C15324T. Others were exclusive to Illumina (most remarkably C11074T, C6990T, C29353T, and C29774T) while others were exclusive to nanopore. Surprisingly dataset C contained more homoplasies than B (Figure 8), of which many are mutations only found in C (e.g. T13402G, A4050C, T13408C, T8022G, C3130T, T28785G). Each of these homoplasies could in principle also be caused by issues with phylogenetic inference, which itself can be affected by homoplasic substitutions.

Figure 8: homoplasies in datasets A, B and C.
Given that B also contains 87 samples from Belgium, this suggests that at least some of the mutation-homoplasies specific to C could be the result of recurrent artefacts. Looking further into some of the most homoplasic of these sites, we see that:
T13402G is a nonsense mutation, and seems to back-mutate 4 times;
T13408C is synonymous, mostly appearing in T13402G mutants; A and G alleles are also observed at the same site 13408 in dataset C.
These two observations further suggest the possibility that selection is not involved in the emergence of these mutations. However, A4050C and T8022G are nonsynonymous.
To test for the possibility that some of these homoplasies might be the result of phylogenetic errors, we masked the most common homoplasies from these datasets one part at a time, to see if other homoplasies would disappear. This had almost no effect.
Another possibility is that these homoplasies might be caused by some of the samples being particularly enriched in recurrent artefacts, rather than artefacts distributed uniformly across all samples. To test this, we compared the sequences of datasets A–C against the full dataset and recorded the minimum number of differences between each sample and any other sample in the dataset. In some sense, this is a measure of the minimum distance of a sample from the rest of the dataset. Most samples have other very small distances (0–1 differences 85% of times for A, 88% for B, 77% for C, median of 0 for A and B and 1 for C). Samples with a distance of at least 3 substitutions were 5% for A and B, and 13% for C. Removing samples with a distance of at least 3 substitutions from A, B or C did not noticeably reduce the number of homoplasies in the corresponding datasets, so it seems like homoplasies and high-distance samples are possibly two separate effects of the same underlying cause.
For future analyses, we filter out all samples with a distance of at least 3 from the rest of the dataset.
Individual noticeable homoplasies
We will now discuss some of the most common homoplasies. As mentioned above, G11083T is the most frequent, appearing 679 times and apparently mutating 21 times forward and 7 times reverting to the original T allele. The T allele is observed in different sequencing technologies and different countries. This is a new non-synonymous (L to F) mutation (ORF1a 3606 nsp6 37) and also is considered one of the best candidates for positive selection ( Notice: this page has been deprecated in favor of a fully redesigned and updated version 2 / Sergei Pond | Observable ), but this homoplasy has also been interpreted in literature as the result of frequent recombination [7]. It appears in all samples from the Diamond Princess cruise ship. Notably the mutation is next to the longest non-terminal homopolymer in the genome, further extending it from 8 consecutive T’s to 10 (Figure 9). The mutation also appears in samples as a within-host polymorphism, as can be seen from the presence of isolated N’s (17 times) and K’s (9 times) in the alignment (see e.g. Figure 9). We also observe this from read files from the Sequence Read Archive (see next section), where the mutation appears as within-patient polymorphism 16 times, and even more often with less stringent filtering and variant calling. When applying more stringent read filtering, the frequency of the T allele seems to consistently decrease. Considering all of these observations, we think that G11083T might be a particularly frequent mutation or artefact. It is unlikely to be the result of positive selective pressure at the amino acid level, as the mutation seems apparently to revert to the original allele many times, and as the same amino acid substitution L→F would also be obtained with the substitution G11083C, which however we never observed in our data.

Figure 9: alignment extract around position 11083.
As a consequence of the observations above, we suspect frequent homoplasies specific to dataset C could be artefacts (or possibly normal mutations that appear as homoplasic due to phylogenetic errors). These include T13402G, which is a nonsense mutation appearing in 51 sequences; its neighbour 13408, in which all three non-reference alleles are observed; and A4050C, a nonsynonymous mutation appearing in 18 sequences. Others are less frequent, and it is particularly unclear if their homoplasy might be caused by phylogenetic errors; these include T8022G (nonsynonymous, appearing in 5 samples), T28785G and C3130T (appearing only in 4 and 3 samples respectively). Recently the dataset C-specific mutations at positions 24389-24390 have been suggested to be strongly affected by local recombination [9], but this might be a phylogenetic artefact caused by other homoplasies.
A pattern observed in many homoplasies (but not those present in C samples specifically) is that they seem to extend pre-existing poly-T runs. The most remarkable example is G11083T above, but the same is observed with its neighbour C11074T (nonsynonymous, not observed within-host, Illumina-specific), with C21575T (nonsynonymous, also observed as within-host polymorphism in a few samples, ubiquitous), and with the Illumina-specific C6990T.
Other frequently mutating homoplasies are C16887T and C15324T (both synonymous not observed within-host), and C6255T (a synonymous variant particularly common in samples from the Netherlands).
Below we show the phylogeny estimated in IQTree after removing all samples with a distance of at least 3 substitutions from the rest of the dataset, and masking the most predominant and dubious homoplasic sites (3130, 3145, 4050, 6990, 8022, 10323, 11074, 11083, 13402, 13408, 14408, 15324, 16887, 21575, and 29353).

Figure 10: IQtree phylogenetic tree after filtering out mutational clusters/recombinations, diverged and isolated samples, and the most common homoplasies.
To test which of these homoplasies might (also) be actual inherited viral mutations, and which are more likely to be non-inherited sequencing artefacts, we measured the phylogenetic signal present in the most homoplasic and/or lab-specific variants using the methods of Borges et al 2018 [10]. First, we masked the most homoplasic sites from the alignment (apparently mutating four times or more) and the homoplasies specific to one lab and mutating at least three times:
187, 241, 335, 1059, 2094, 3037, 3130, 3145, 4050, 6255, 6990, 8022, 8782, 9223, 10323, 10741, 11074, 11083, 11704, 13402, 13408, 14408, 14724, 14786, 14805, 15324, 16887, 17247, 19684, 20148, 21137, 21575, 23403, 24034, 24378, 25563, 26144, 26461, 26681, 27384, 28077, 28826, 28854, 29353, 29700, 29736.
These sites were determined iteratively, first removing the most homoplasic sites, repeating the phylogenetic inference, and then again removing the most homoplasic sites.
We then inferred a phylogeny from the masked alignment using IQTree. Finally, we measured the phylogenetic signal of each of the homoplasies listed above by coding the observed allele as a categorical variable and using the delta statistic. The delta statistic uses Shannon entropy to measure the degree of phylogenetic signal between traits and phylogenies: the idea is that the better a phylogeny is associated with a given trait, the better it is able to retrace trait’s evolution (or in probabilistic terms, to infer the ancestral states with minimal uncertainty). Polytomies in the phylogeny were randomly split to obtain a bifurcating tree. Most of the homoplasies, in particular those appearing in only a few sequences, show close to no phylogenetic signal (Figure 11), supporting the hypothesis that they are artificial. On the other hand, many homoplasies, including site 11083, show strong phylogenetic signal, suggesting that they originated, at least once, as a true mutational event. Of course, this analysis has substantial limitations, it ignores uncertainty in the tree and noise from possible artefacts within the remaining sites. It also does not tell us if a variant is both a true mutational event as well as the result of recurrent sequencing biases, as possible for position 11083.
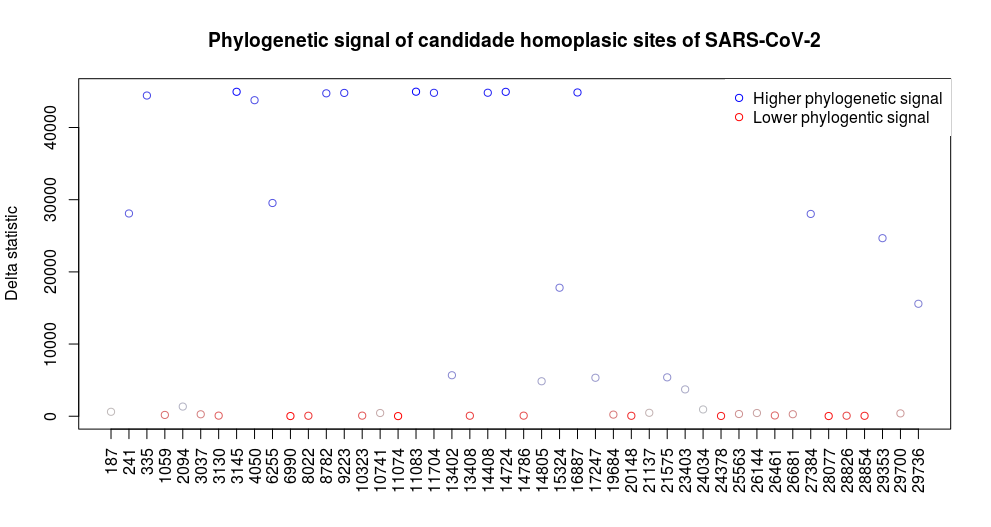
Figure 11: Phylogenetic signal of candidate homoplasic sites using the method of Borges et al [10].
Within-sample variation
A possible way to include more genetic variation data and to investigate possible artefacts is to consider within-host variation from sequencing read files. We downloaded the available 450 SARS CoV 2 Illumina sequencing FASTQ files from the Sequence Read Archive (SRA, Home - SRA - NCBI ) on 12th April 2020. The pipeline we used for processing the reads in the within-sample variation section is available at GitHub - conorwalker/covid19 . From preliminary analyses of the 8 sequencing datasets of Shen et al 2020 [11] we realized that in order to remove most variants that might emerge from mapping and sequencing artefacts, stricter filters than usual were needed. More specifically, we used fastp (GitHub - OpenGene/fastp: An ultra-fast all-in-one FASTQ preprocessor (QC/adapters/trimming/filtering/splitting/merging...)) to:
- trim adapters and polyX tails from reads
- trim 15 bases from the start and end of each read
- only retain reads with high quality sequence (≥Q35) at ≥90% of the read length
- remove reads with low quality (<Q30) at the 3’ end
- only retain reads of length ≥50 before alignment with bwa-mem (GitHub - lh3/bwa: Burrow-Wheeler Aligner for short-read alignment (see minimap2 for long-read alignment)), possible PCR duplicates were removed using MarkDuplicates from Picard tools (GitHub - broadinstitute/picard: A set of command line tools (in Java) for manipulating high-throughput sequencing (HTS) data and formats such as SAM/BAM/CRAM and VCF.), and any reads aligned through hard or soft clipping were removed from the bam file.
We only consider variants at a site if the depth of coverage is ≥5 and the base quality is ≥30.
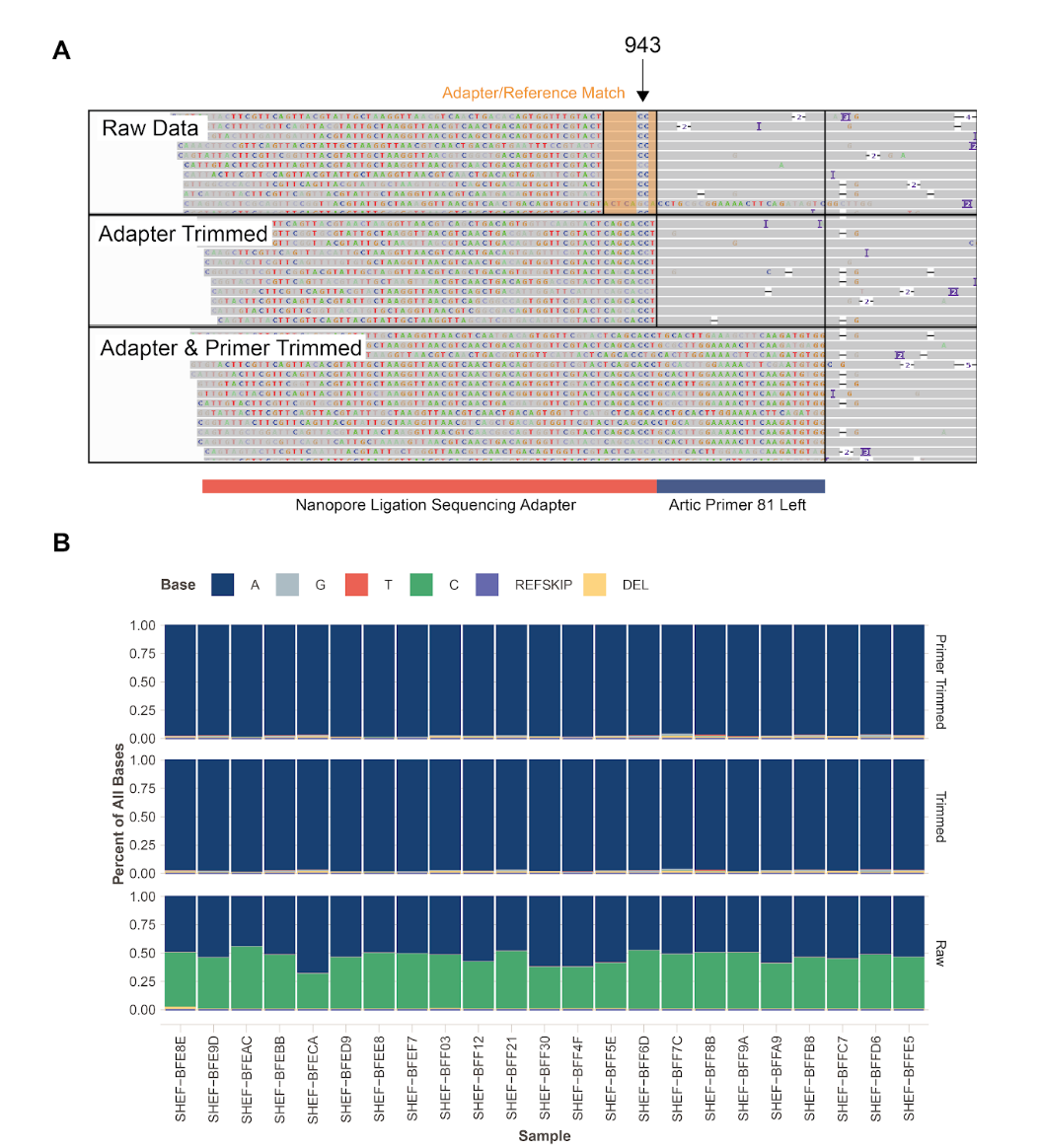
The strong filters of removing 15bp from each read end and removing clipped reads were useful because some variants were observed at very high frequency in some samples but only at read ends or only in clipped reads. For example, at reference position 10779, an A/T polymorphism was observed in 7 out of 8 samples from Shen et al 2020, usually with A being the minor allele, and once being the majority allele. In the 8th sample, allele A appeared fixed. We observed allele A only in read ends (see Figure 12), and after trimming read ends this polymorphism was no longer detected. This suggests that recurrent artefacts might be present not only at the level of substitutions between consensus sequences, but also as frequent, apparent within-host variants.

Figure 12: extract of a bam file from a read alignment of a single sample from Shen et al [11]. Position 10779 appears polymorphic, but the minority variant is only observed near read ends.
Generally, most samples contain 0–5 variants (median 1, mean 8.2). However, some samples have many more, reaching 564 and 433 in two cases (SRR11494637 and SRR11494664 respectively). Coverage or mixed infection/contamination do not seem the issues. Instead, these samples were mostly made of clipped reads, i.e. reads that only partly map on the reference genome. Removing these reads eliminated these extreme cases (Figure 13), but other samples with extreme numbers of variants can still be found, for example SRR11494662 with 359 within-host variants. As samples with an extreme number of within-host variants persist, a more rigorous filtering procedure might still be required before we can confidently interpret within-host variant calls. We therefore offer a word of caution when attempting to interpret the results of such variant calling methods, and minimally recommend a stringent set of filters (as outlined above), as well as removing samples with more than 2% of “N”s within reads.
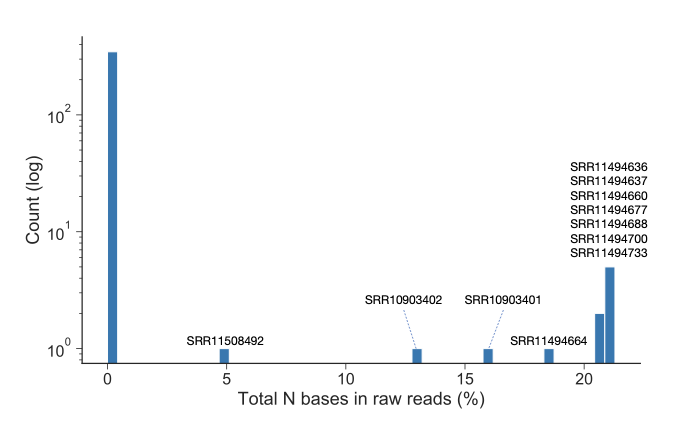
Figure 13: Histogram of the number of Illumina-sequenced samples (Y axis) with a given percentage of “N” within its reads (X axis). We filtered out samples with more than 2% (all those whose name is shown in the plot).
The mutational spectrum of within-host variants seems extremely shifted toward G→T variants, even more than when comparing consensus sequences (Figure 6) suggesting that most of these variants might be the result of Illumina sequencing errors and/or sequencing biases, or otherwise that most of these new G→T mutations do not reach fixation due to selective forces. We have not yet investigated possible nanopore sequencing biases from within-host variation data.
Within-host variation at homoplasic sites
Most homoplasies found only in the consensus sequences rarely appear polymorphic within-host, and they do not show evident signs of mapping or calling issues. G11083T is again exceptional in this regard. This variant is polymorphic in many samples, as mentioned above, and the frequency of the variant T allele seems to decrease with stricter read filtering. Furthermore, this position is also associated with strange deletion patterns (Figure 14), with a 1bp deletion being present in many reads at this position despite this being within a coding sequence. We also found the same pattern at the neighbouring position 11074 for sample SRR11494457 (Figure 15). Here coverage is low, the derived allele T is the higher frequency allele, and a similar deletion pattern to position 11083 is observed. It is not clear to us if these observations suggest that sequencing errors might be common at this position due to the poly-T. We plan to investigate nanopore read data to further look into this issue.
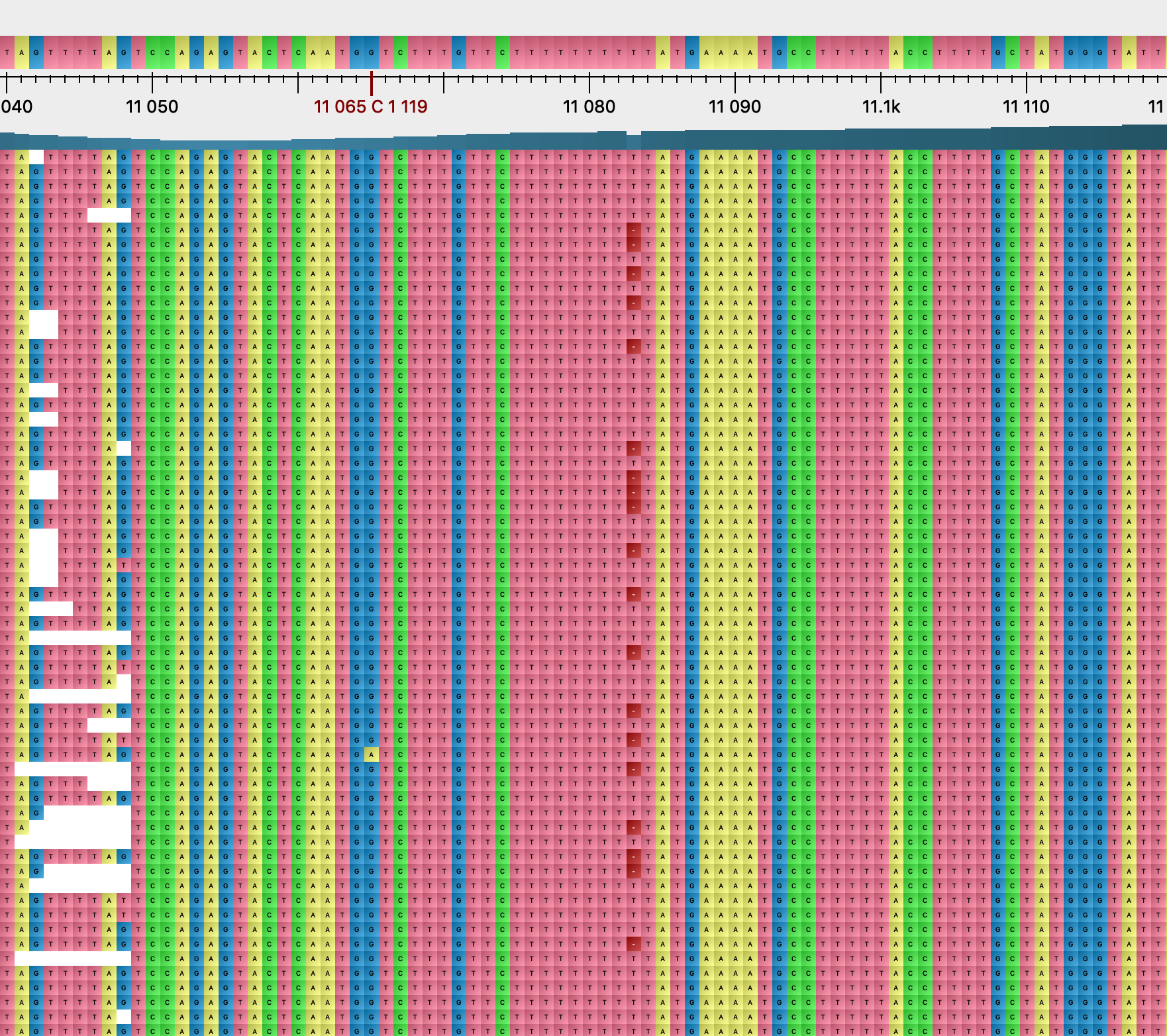
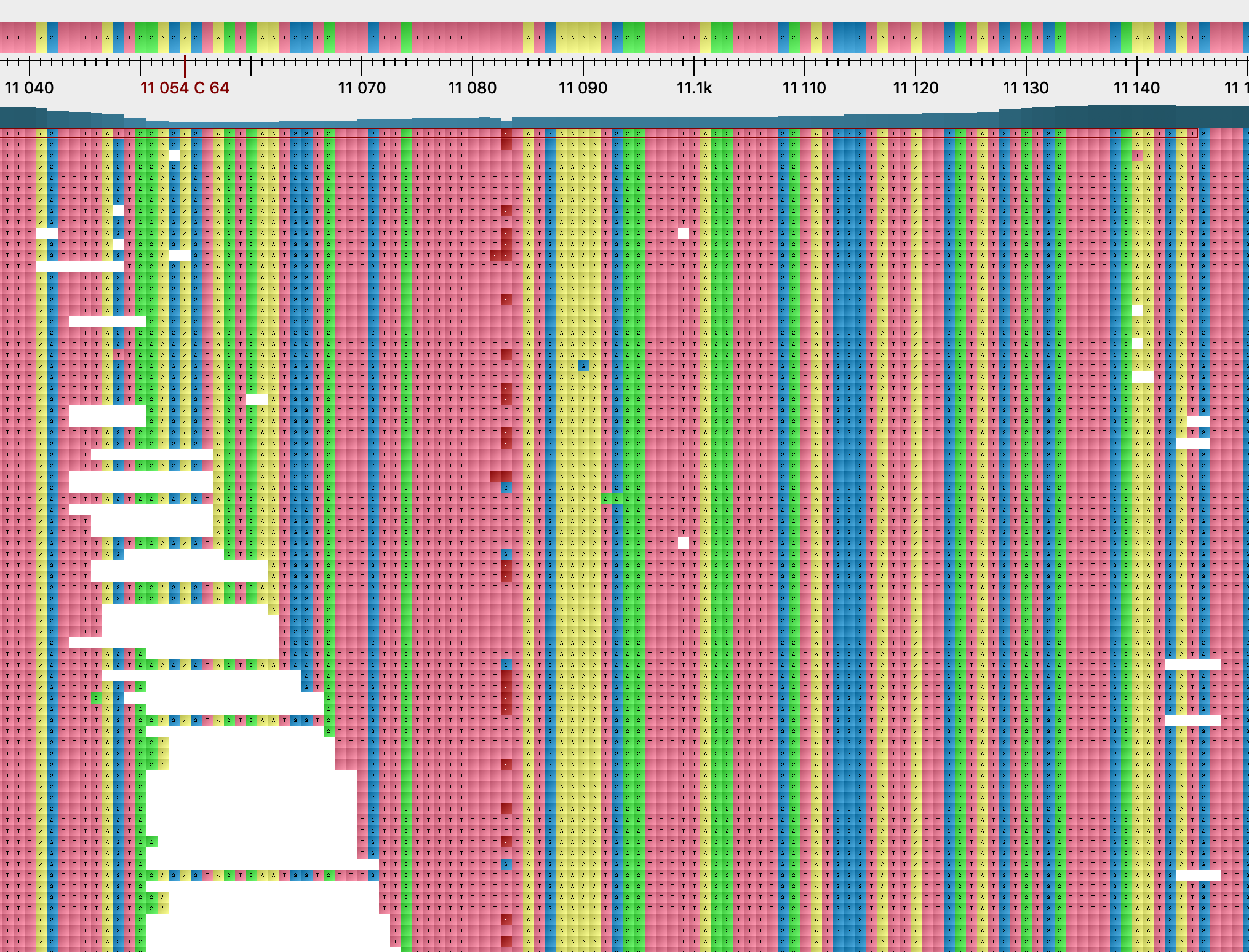
Figure 14: two read alignments near position 11083. Dark red squares represent deleted bases. Top: sample SRR11397719. Bottom: sample SRR11494651.
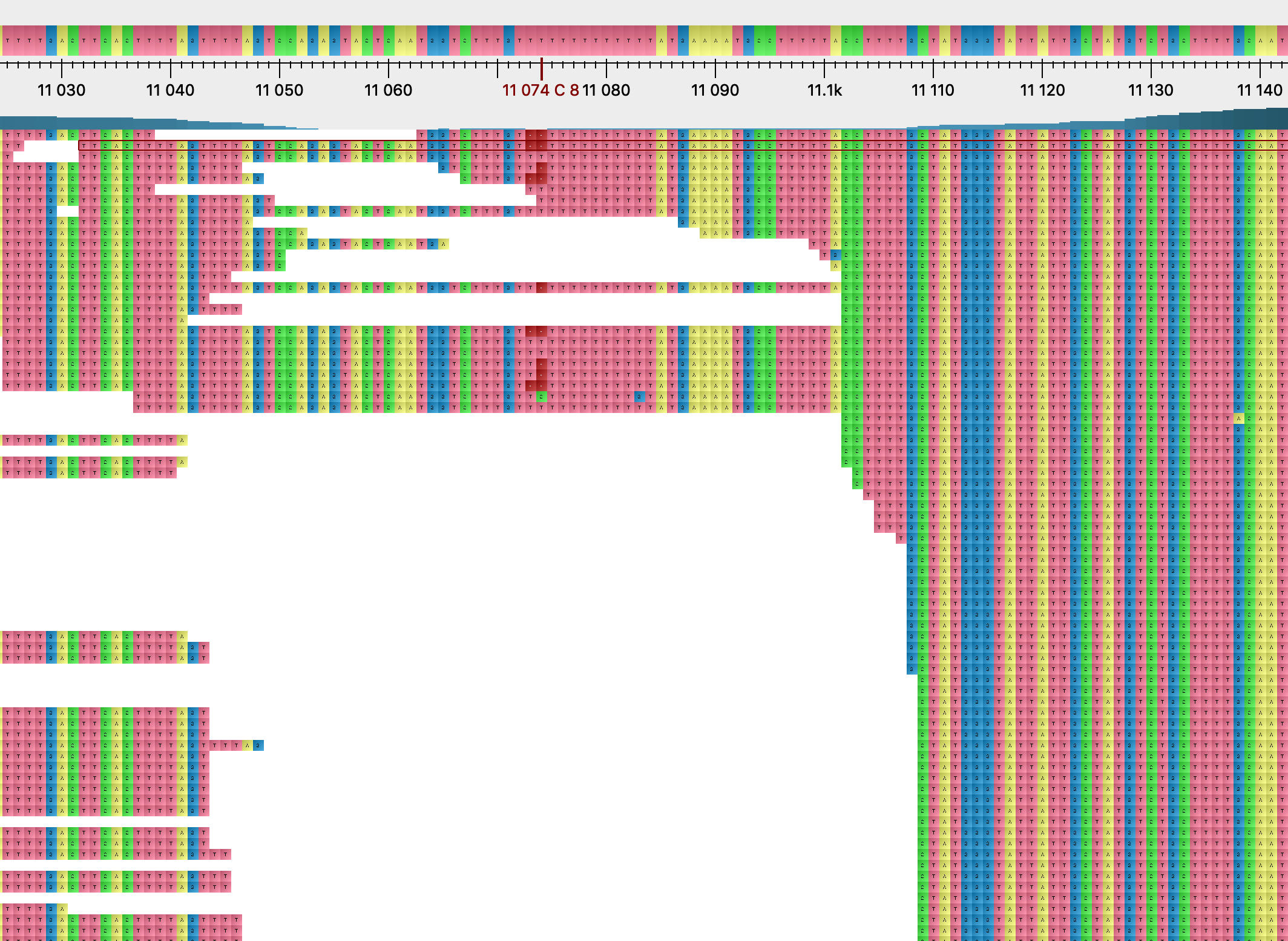
Figure 15: read alignment near position 11074, sample SRR11494457.
Acknowledgements
We would like to thank all the authors who have kindly deposited and shared genome data on GISAID (https://www.gisaid.org/) and in particular Piet Maes for helpful suggestions. A full table of acknowledgments for GISAID contributors can be found here: ProblematicSites_SARS-CoV2/data/gisaid_hcov-19_acknowledgements.tsv at master · W-L/ProblematicSites_SARS-CoV2 · GitHub . We also thank Andrew Rambaut, Ewan Birney, Umberto Perron and Graham Jones for helpful discussions.
References
[1] Shu, Yuelong, and John McCauley. “GISAID: Global initiative on sharing all influenza data–from vision to reality.” Eurosurveillance 22.13 (2017).
[2] Katoh, Kazutaka, and Daron M. Standley. “MAFFT multiple sequence alignment software version 7: improvements in performance and usability.” Molecular Biology and Evolution 30.4 (2013): 772-780.
[3] Minh, Bui Quang, et al. “IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era.” Molecular Biology and Evolution 37.5 (2020): 1530-1534.
[4] Didelot, Xavier, and Daniel J. Wilson. “ClonalFrameML: efficient inference of recombination in whole bacterial genomes.” PLoS Computational Biology 11.2 (2015).
[5] Sagulenko, Pavel, Vadim Puller, and Richard A. Neher. “TreeTime: maximum-likelihood phylodynamic analysis.” Virus Evolution 4.1 (2018): vex042.
[6] Meacham, Frazer, et al. “Identification and correction of systematic error in high-throughput sequence data.” BMC Bioinformatics 12.1 (2011): 451.
[7] Yi, Huiguang. “2019 novel coronavirus is undergoing active recombination.” Clinical Infectious Diseases: An Official Publication of the Infectious Diseases Society of America (2020).
[8] https://doi.org/10.1101/2020.04.29.069054
[9] Hadfield, James, et al. “Nextstrain: real-time tracking of pathogen evolution.” Bioinformatics 34.23 (2018): 4121-4123.
[10] Borges, Rui, et al. “Measuring phylogenetic signal between categorical traits and phylogenies.” Bioinformatics 35.11 (2019): 1862-1869.
[11] Shen, Zijie, et al. “Genomic diversity of SARS-CoV-2 in coronavirus disease 2019 patients.” Clinical Infectious Diseases (2020).
[12] Forster, Peter, et al. “Phylogenetic network analysis of SARS-CoV-2 genomes.” Proceedings of the National Academy of Sciences 117.17 (2020): 9241-9243.
[13] https://doi.org/10.1101/2020.04.26.062422