Updated analysis with data from 17th May 2020
Nicola De Maio, Conor Walker, Nick Goldman
European Molecular Biology Laboratory, European Bioinformatics Institute, Hinxton, Cambridgeshire, United Kingdom.
Summary
After our initial post, we worked to improve our analyses. First, we included the new genomes and the metadata available from GISAID on 17th May 2020. Then, following the finding of recurrent sequencing artefacts at positions 24389-24390, we masked these positions, and we paid more attention to variants that are in proximity to each other and linked: such sequencing errors would have more potential to cause topological errors and thus avoid detection and cause misinterpretation of other variants as homoplasic. We also used information from frequent ambiguous bases present in multiple consensus sequences within the same alignment column. These ambiguities usually mark positions that show multiple alleles within the same sample, and therefore can be indicators of recurring within-host mutations or recurring sequencing artefacts.
The new masking recommendations are summarized in our VCF file: see our separate post Masking strategies for SARS-CoV-2 alignments , the VCF file itself https://github.com/W-L/ProblematicSites_SARS-CoV2/blob/master/problematic_sites_sarsCov2.vcf , and the same file in human readable format https://github.com/W-L/ProblematicSites_SARS-CoV2 .
Highly homoplasic sites
We still advise masking positions 11074, 11083, 16887, 21575 when inferring a phylogeny or testing for positive selection, as it is not clear if variants at these positions emerge from true frequent mutations or from sequencing artefacts. However, because they are highly homoplasic (appearing to emerge many times independently) they have the potential to bias phylogenetic inference, while providing only limited phylogenetic signal.
Sites with ambiguous characters
We find sites that have high proportions of ambiguity characters while also showing patterns of variants and ambiguity that are specific to one country or sequencing lab. The most extreme of these positions are 19484 where 222 ambiguities were observed, and 8022, where 62 ambiguous characters were observed. We suggest masking these positions, because while some level of within-host polymorphism is expected due to within-host mutations, mixed infections and contamination, a high number of ambiguities (in particular at sites with low diversity) can be a sign or recurring mutations or sequencing artefacts.
The full list of sites we recommend masking because of ambiguous characters is:
- 635 (9 variant bases and 6 ambiguous bases, found particularly in UK sequences by COG-UK)
- 2091 (9 variant and 6 ambiguous, enriched from India, NCDC)
- 2094 (6 variant and 6 ambiguous, enriched from Belgium KU Leuven and India NCDC)
- 3564 (46 variant and 15 ambiguous, enriched from Australia VIDRL)
- 6869 (4 variant and 12 ambiguous, enriched from Italy Caporale)
- 8022 (9 variant and 62 ambiguous, enriched from India NCDC, Belgium KU Leuven)
- 8790 (11 variant and 6 ambiguous, enriched from Australia VIDRL)
- 10129 (2 variant and 10 ambiguous, enriched from USA California Chan-Zuckerberg Biohub )
- 13476 (5 variant and 5 ambiguous, enriched from Turkey Ministry of Health)
- 13571 (6 variant and 14 ambiguous, enriched from Australia VIDRL)
- 15922 (3 variant and 12 ambiguous, enriched from USA California Chan-Zuckerberg Biohub )
- 19484 (16 variant and 222 ambiguous, enriched from UK COG-UK)
- 22335 (3 variant and 11 ambiguous, enriched from UK COG-UK)
- 24933 (39 variant and 16 ambiguous, enriched from Australia VIDRL).
Homoplasic sites common within one sequencing lab
We also re-ran the analysis of homoplasic sites described in our original post but this time making use of the new data and focusing on variants that show high levels of homoplasy relative to their prevalence, and that are mostly found in samples sequenced from a single lab; these variants have a high probability of being sequencing artefacts rather than the result of recombination, contamination, hypermutability or positive selection, and so masking them should reduce the topological distortion caused by artefacts while sacrificing less true phylogenetic signal. We recommend masking the following sites:
- 3145 (10 variant, enriched from Netherlands Erasmus)
- 4050 (8 variant, enriched from Belgium KU Leuven)
- 5736 (6 variant, enriched from Turkey Ministry of Health)
- 11535 (22 variant, enriched from Australia VIDRL)
- 13402 (27 variant, enriched from Belgium KU Leuven)
- 13408 (3 variant, enriched from Belgium KU Leuven, linked with 13402)
- 14277 (9 variant, enriched from Australia VIDRL)
- 24389 (5 variant, enriched from Belgium KU Leuven)
- 24390 (5 variant, enriched from Belgium KU Leuven)
- 26549 (12 variant, enriched from Turkey Ministry of Health)
- 29037 (5 variant, enriched from Australia VIDRL) TCT->TTT
- 29553 (259 variant, enriched from USA Washington UW or “Seattle Flu Study”)
We observe an enrichment of GGT to GTT substitutions in homoplasic sites specific to the Australian VIDRL (2 out of 3, sites 11535 and 14277) and sites with ambiguities specific to the Australian VIDRL (4 out of 4, sites 3564, 8790, 13571, and 24933). This is very different from the overall mutational trend, where most variants are C to T, but it resembles the G to T skew we observed in within-host variants in our original post. It’s not clear to us if this pattern is a result of RNA degradation, RNA modification, some other biological process, or specific aspects of the chemistry and bioinformatics used by the lab (do you have suggestions @torsten ?), either way we suspect this observation might help to improve the calling of consensus sequences, or to understand and account for SARS-CoV-2 hypermutable sites.
Neighbour linked variants
Finally, we investigated linked, proximal variants (<10bp apart). These, if recurrent sequencing artefacts as for the case of 24389-24390, have the potential to cause substantial phylogenetic errors and bias in downstream analyses (including our own inference of homoplasy). We still mask linked sites we previously identified as problematic (24389-24390, 11074 and 11083, 13402 and 13408; see above), and then we investigated the most prevalent of these apparent mutational patterns, 28881-28883 (see our original post). We found no evidence of possible sequencing artefacts at these positions from read data, and we found that almost all samples with mutations at 28881-28883 cluster closely together (see e.g. Figure 1); this is true also after masking some of these positions. The few exceptions are probably just rare cases of contamination or recombination. We conclude that, apart from a few possible samples where the minor variant at 28881-28883 was acquired through recombination or contamination, 28881-28883 likely represents a true, single multi-nucleotide mutational event, and we do not recommend masking these positions anymore. The only exception might be in phylodynamic analysis, where it might make sense to mask only 2 out of these 3 sites to account for the fact that these likely result from a single mutational event.
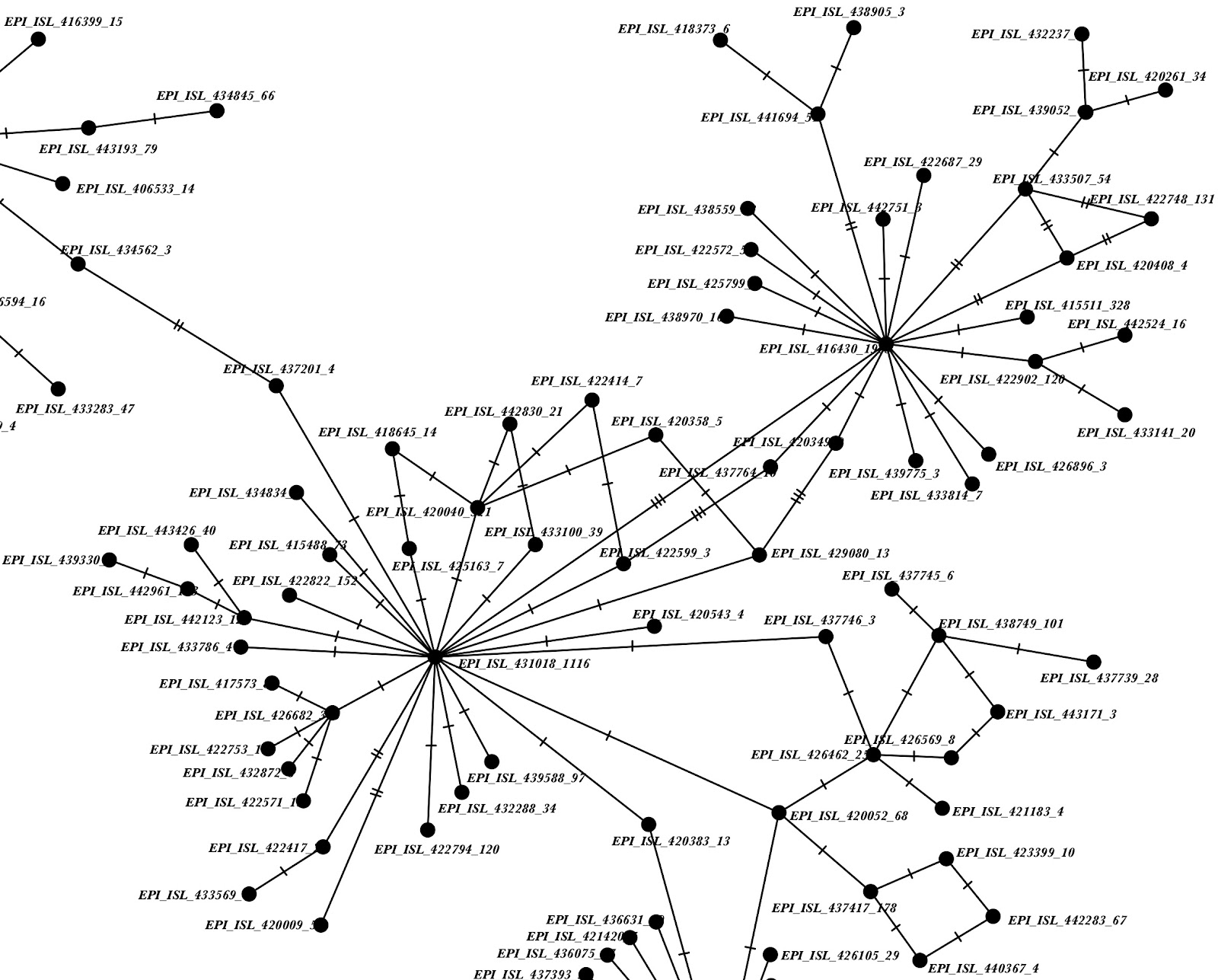
Figure 1: extract of a phylogenetic network obtained from SARS-CoV-2 sequences in POPART [1]. This is an extract of a simplified network obtained by considering only high-frequency variants (those appearing in at least 50 sequences). The top right corner contains all the sequences with variants at positions 28881-28883 (notice the three edges with 3 substitutions separating these sequences from the rest of the network). Node names show the identifier of one of the sequences with the given haplotype, followed by the number of such sequences. Nodes representing fewer than 3 sequences have not been included.
Other linked, proximal variants were observed at a much lower prevalence, and so we decided to mask these (only in the samples which carried them) to prevent possible biases while only sacrificing limited phylogenetic signal; it would make sense to further investigate them individually in the future, as we did for 28881-28883.
Some of the most remarkable of these sites (due to involving at least 3 nearby perfectly or almost perfectly linked substitutions and sometimes from a single sequencing laboratory) were:
- 6971, 6975, 6977 (found in 6 samples from Australian VIDRL)
- 13512, 13513, 13514 (found 2 samples)
- 13686, 13687, 13693 (found 2 samples)
- 21302, 21304, 21305 (found in 3 samples)
- 27382-27384 (found in 4 samples)
- 28004-28006, -280089 (found in 3 samples from Luxemburg)
We also think it is worth further investigating positions 6309, 6310 and 6312 (and of these in particular 6310), as these form a cluster of nested variants showing some similarity with the 11074-11083 cluster (in this case these variants extend a poly-A rather than a poly-T).
Other changes
We made other changes to our analyses that do not affect our proposed masked sites, but that affect the alignment we work with. In particular, we now mask the first and last 30 non-ambiguous non-gap characters of each sequence, in order to mask most alignment errors without automatically masking large regions at the ends of the genome. We also mask 7bp surrounding missing parts of each sequence, again to avoid errors attributable to alignment and low coverage.
Recombination and contamination
We further investigated the possible presence of recombination using a phylogenetic network approach (see Figures 1 and 2). We found no clear evidence of recombination; however, there are many loops in the network. After removing highly homoplasic sites, we still observe many of these loops, which do not seem to be caused by highly homoplasic mutations. Because these loops seem to be present mostly in the terminal parts of the network and involve sequences with very few representatives, it seems most likely that these are the results of contamination, where the genomes of two different strains contribute to a single consensus sequence (but we cannot exclude recombination events near terminal nodes that would result in the same observed patterns). Contamination/recombination not only creates topological uncertainty, but is likely to cause biases in estimation of mutation rate, homoplasy, and selection.

Figure 2: Extract of phylogenetic network (see Fig. 1 for details). In this area of the network, several loops can be noticed, despite this representing a very simplified version of the full network.
Acknowledgements
We would like to thank all the authors who have kindly deposited and shared genome data on GISAID (https://www.gisaid.org/). A full table of acknowledgments for GISAID contributors can be found here: https://github.com/W-L/ProblematicSites_SARS-CoV2/blob/master/data/acknowledgements_GISAID.zip
References
[1] Leigh, Jessica W., and David Bryant. “POPART: full‐feature software for haplotype network construction.” Methods in Ecology and Evolution 6.9 (2015): 1110-1116.