[Update information - Genomic surveillance of the state of Rio de Janeiro, Brazil, in the period between August, 04th, 2021 and August, 16th, 2021]
Since last update, we have sequenced new 377 SARS-CoV-2 genomes from the state of Rio de Janeiro, Brazil, as part of the Genomic Surveillance Program of the Rede Corona-ômica-RJ project. We have sampled, so far, 3,952 genomes from the state. As expected, the Delta variant (lineages B.1.617.2, AY.4, AY.5, AY.6, AY.7.1, AY.7.2, AY.12, AY.20 and AY.25) has become dominant in Rio de Janeiro and now corresponds to ~89.2% of the samples collected in August. We are investigating if such high lineage diversity within Delta in Rio de Janeiro is originated by new introductions in Brazil or a misclassification by PANGO-learn. While Gamma variant is still present in the sample (10.8%), Alpha has completely disappeared. No other lineages were identified in the sample either.
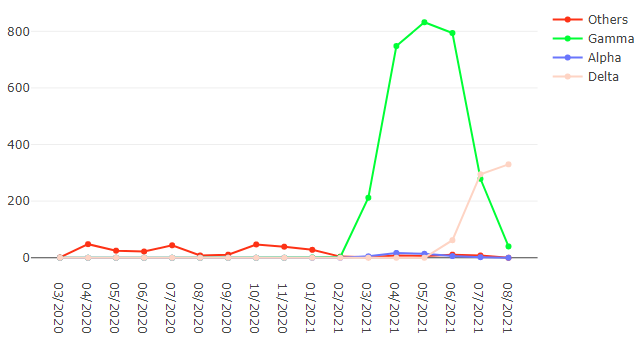
Figure 1. Frequency of SARS-CoV-2 variants of concern in the state of Rio de Janeiro, Brazil.