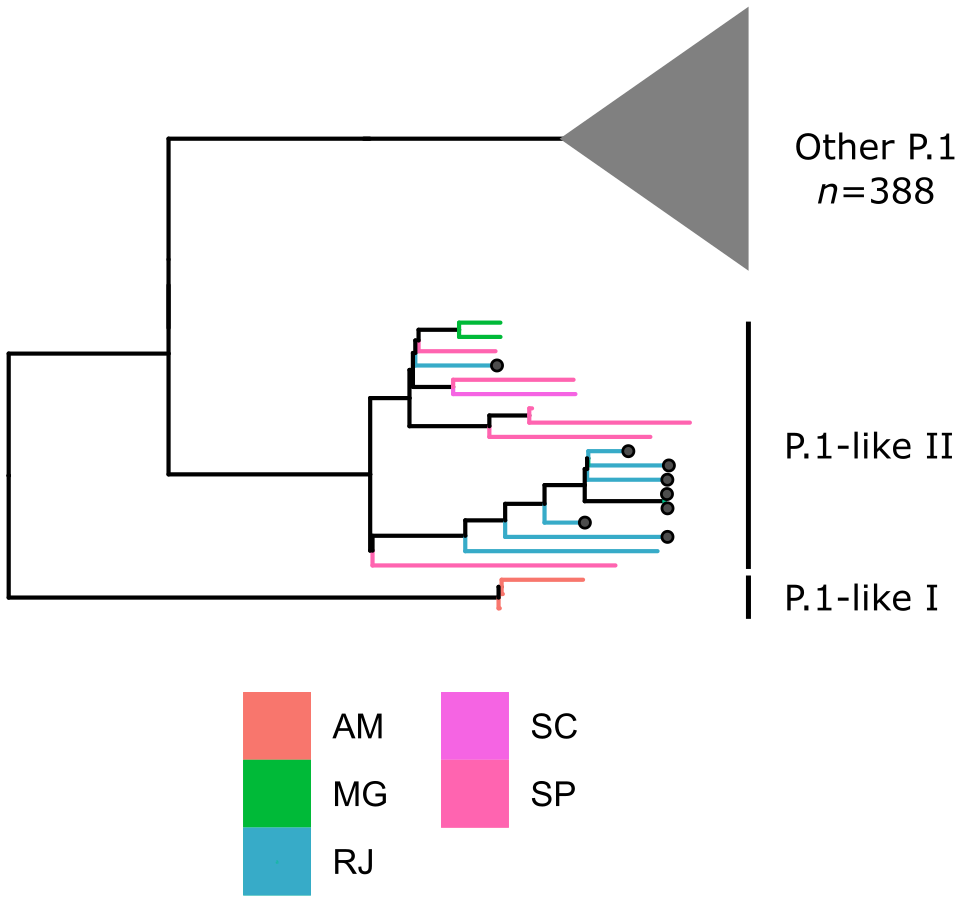Genomic Surveillance of SARS-CoV-2 in the State of Rio de Janeiro, Brazil: a technical briefing
Luiz G P de Almeida¹, Alessandra P Lamarca¹, Ronaldo da Silva F Jr¹, Liliane Cavalcante¹, Alexandra L Gerber¹, Ana Paula de C Guimarães¹, Douglas Terra Machado¹, Cassia Alves², Diana Mariani², Thais Felix Cruz², Mario Sergio Ribeiro³, Silvia Carvalho³, Flávio Dias da Silva4, Marcio Henrique de Oliveira Garcia4, Leandro Magalhães de Souza5, Cristiane Gomes Da Silva5, Caio Luiz Pereira Ribeiro4, Andréa Cony Cavalcanti5, Claudia Maria Braga de Mello³, Amilcar Tanuri²ª, Ana Tereza R Vasconcelos¹ª*
ªBoth authors coordinated this work
*Corresponding author : [email protected]
1 Laboratório de Bioinformática, Laboratório Nacional de Computação Científica, Petrópolis, Brazil.
2 Departamento de Genética, Instituto de Biologia, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil.
3 Secretaria Estadual de Saúde do Rio de Janeiro, Rio de Janeiro, Brazil.
4 Secretaria Municipal de Saúde Rio de Janeiro, Rio de Janeiro, Brazil
5 Laboratório Central de Saúde Pública Noel Nutels, Rio de Janeiro, Brazil.
The Genomic Surveillance Program of the state of Rio de Janeiro is part of a task force composed by the Secretary of Health from the state of Rio de Janeiro, the Secretary of Health from the city of Rio de Janeiro, the Noel Nutels Central Laboratory (LACEN-RJ) and the Corona-Ômica-RJ Network through partnership with the Bioinformatics Laboratory of the National Laboratory for Scientific Computing (LNCC) and the Laboratory of Molecular Virology (LVM) at the Federal University of Rio de Janeiro (UFRJ).
This project is funded by the Research Support Foundation of the State of Rio de Janeiro (FAPERJ). The Program integrates data related to the viral genetic alterations, transmission and the course of infection among cities in Rio de Janeiro. This report aims to: (i) present the epidemiological situation of the SARS-CoV-2 genomic surveillance at the state levels, and (ii) describe the emergence of possible new mutations and viral variants.
Genomic Epidemiology
The state of Rio de Janeiro, Brazil, contains the third largest city of South America, which is further expanded into a metropolitan area containing multiple industrial cities. Hence, it is not only common for workers to commute daily between neighbouring cities, but Rio de Janeiro also receives a high number of interstate corporate travels. The large population coupled with this daily/seasonal travelling dynamics favours the introduction and spread of new SARS-CoV-2 lineages in the state.
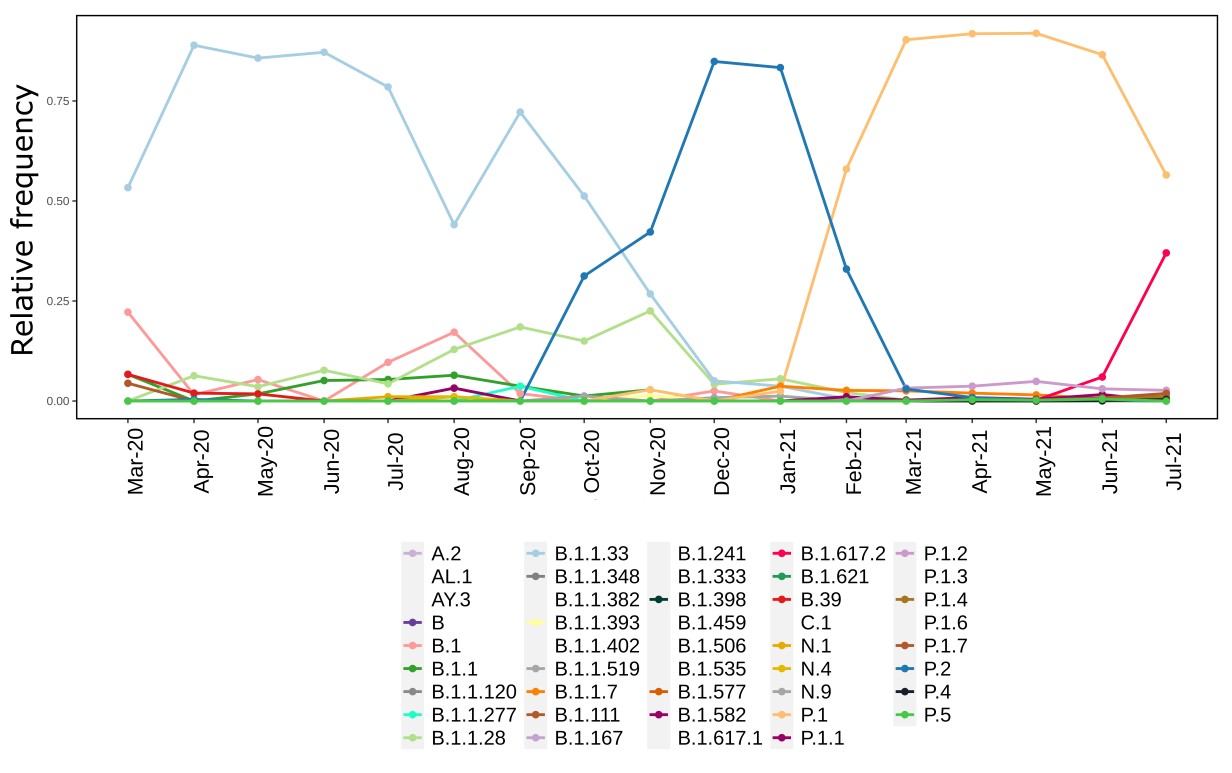
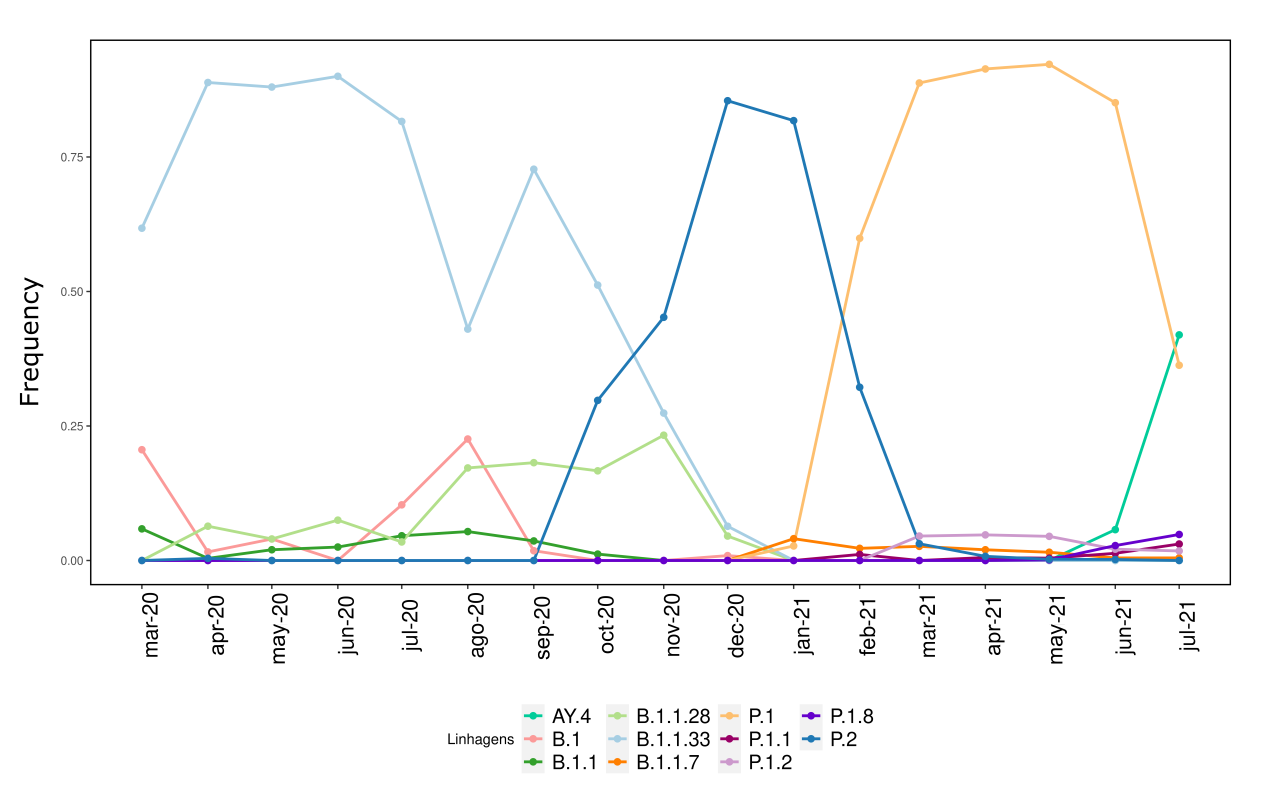
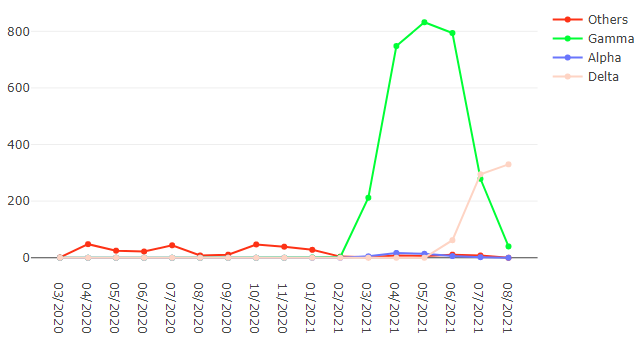
We analysed in both spatio-temporal and phylogenetic contexts 90 SARS-CoV-2 samples collected between March 24, 2021 and March 28, 2021 from 20 municipalities of the state of Rio de Janeiro. Previous sequencing of SARS-CoV-2 samples from the state available in GISAID [1–3] indicated that the B.1.1.33 lineage had a high prevalence from April to October, 2020. P.2 lineage was first detected in September, 2020 and by October had already substituted B.1.1.33 as the prevalent lineage in the state. P.2 gradually increased its frequency until February, 2021, when it reached 64% of samples sequenced (Figure 1). By January, 2021 there was a fast replacement of P.2 by P.1 lineage, the latter being present in 94% of the sampling sequenced for this report (March 24, 2021 to March 28, 2021; Figure 1 and Figure 2A). Currently, P.1 is the dominant lineage widespread across all regions in the State (Figure 2B-C).
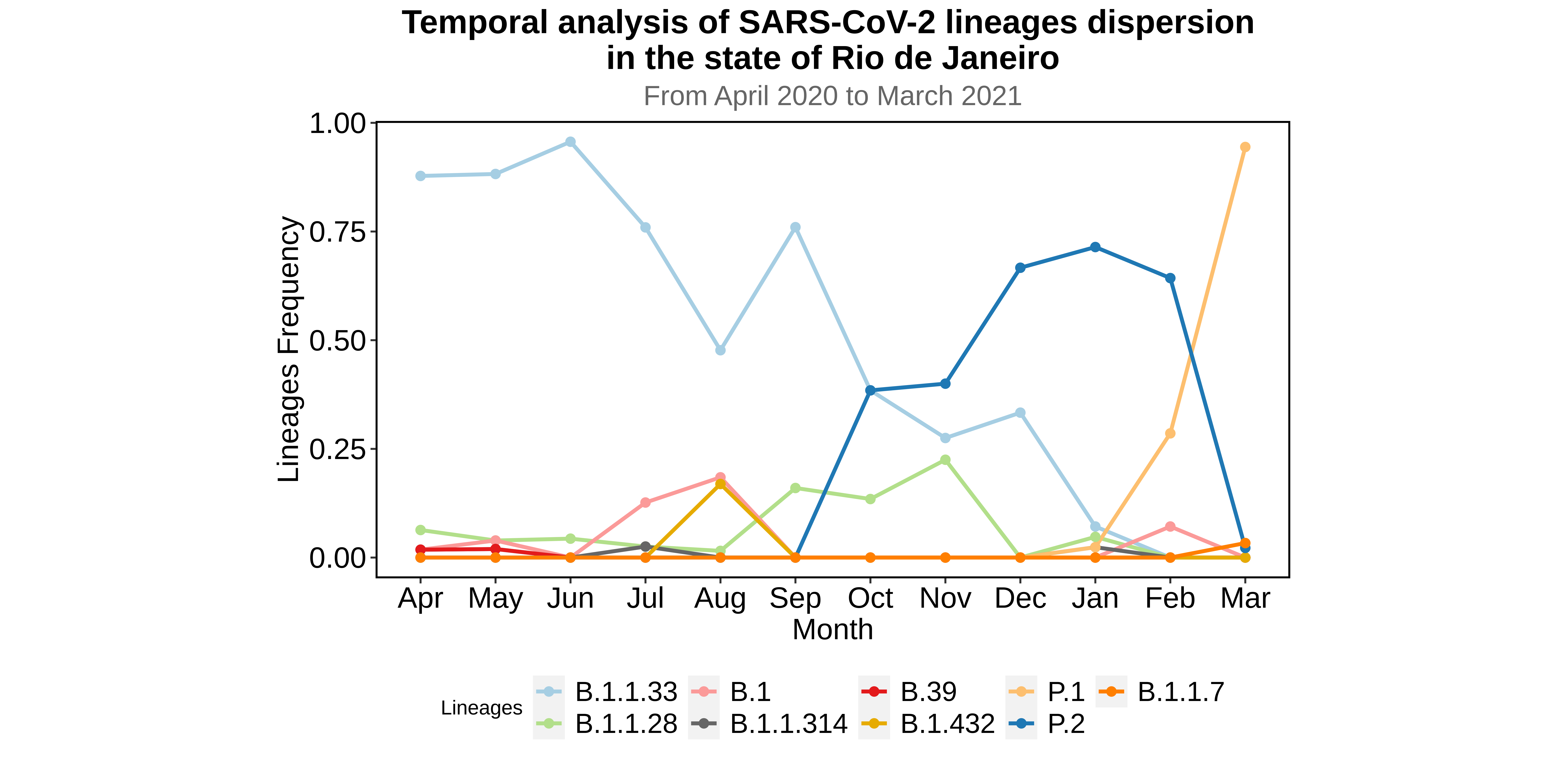
Figure 1. Dispersion of SARS-CoV-2 lineages from April 2020 to March 2021 in the state of Rio de Janeiro.
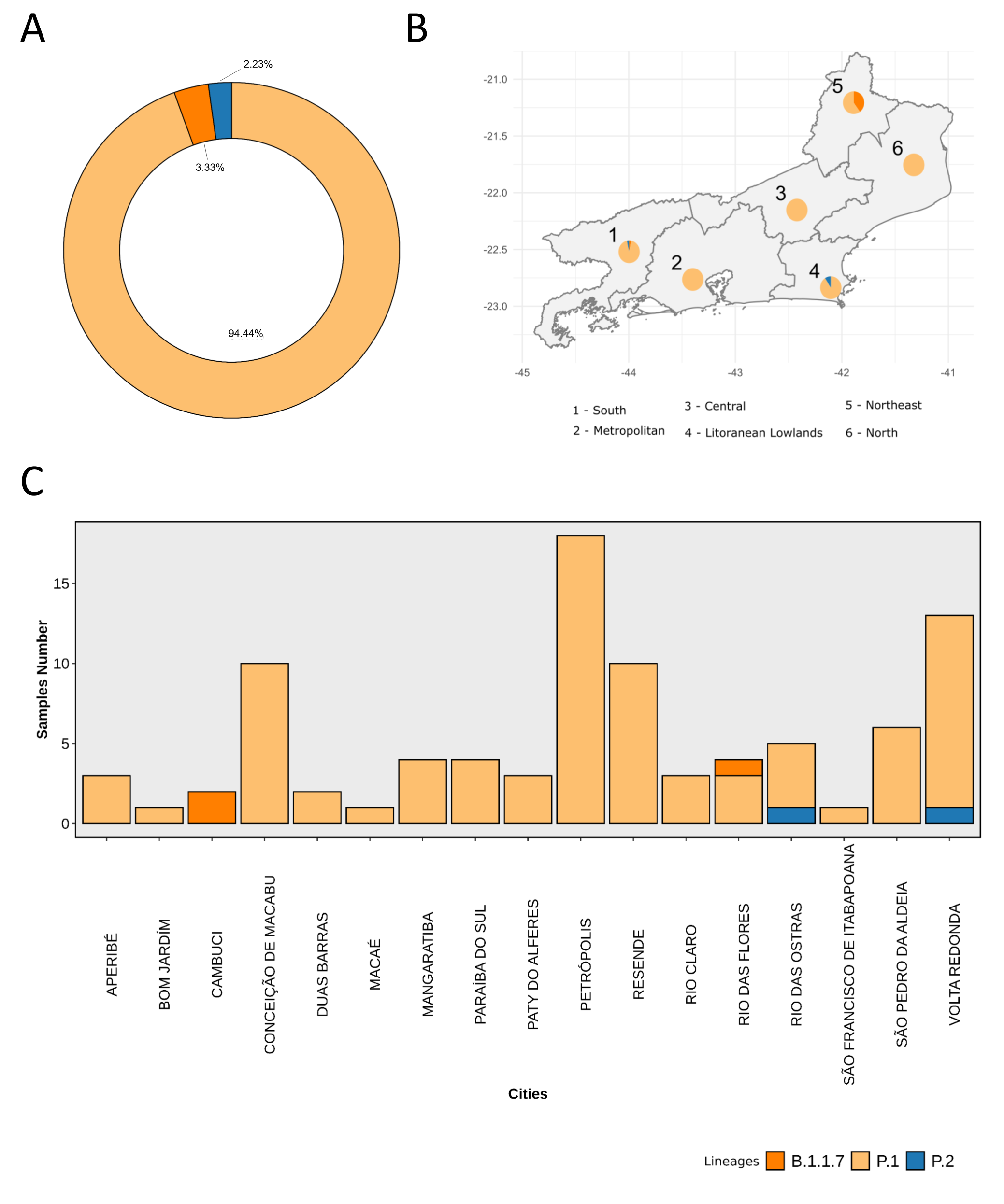
Figure 2. Frequency and spatial analysis of SARS-CoV-2 lineages found in samples from March 2021. A) Donut plot showing the total proportion of lineages identified in the 90 samples sequenced in March 2021. B) Proportion of lineages in each macro-region of the state of Rio de Janeiro. C) Proportion of lineages across the 17 cities investigated in this study.
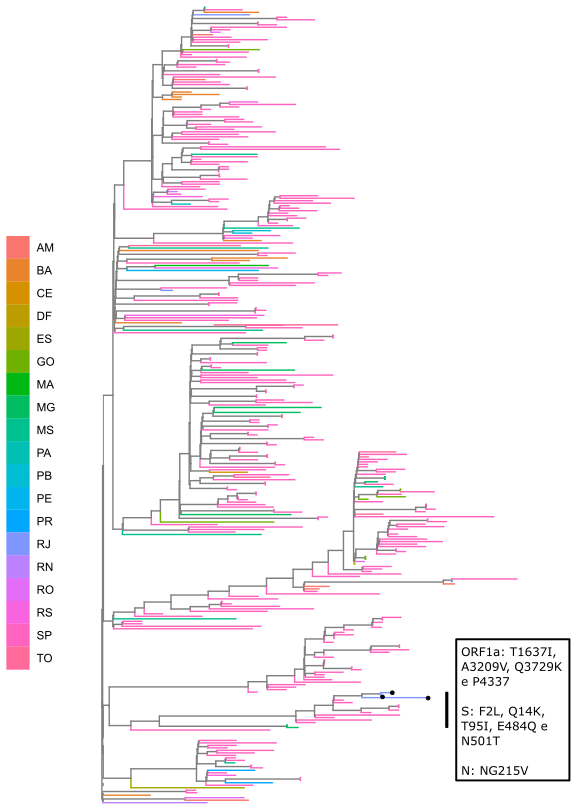
Local transmission of a previously undefined cluster of P.1 lineage was identified in the municipality of Conceição de Macabu in the North region of the state Local transmission of a previously undefined cluster of P.1 lineage was identified in the municipality of Conceição de Macabu in the North region of the state. This cluster contains up to nine mutations in addition to the P.1 lineage-defining ones: four on ORF1ab (synC1150T, synC1912T, D762G, T1820I), two on ORF3a (D155Y,S180F), one on the M protein (synC26954T), one on the N protein (synC28789T) and one on the S protein (A262S) (Figure 3, black dots in tree). We observed that these sequences formed a monophyletic clade with samples from the states of Rio Grande do Sul and São Paulo in Brazil, Spain, Netherlands, Australia and the United States, which consistently bears five of these mutations (synC1912T, D762G, T1820I, D155Y, synC28789T). The mutation D155Y was proposed to lower the viral affinity to the host’s caveolin-1 protein, avoiding cell apoptosis and extending the infection’s asymptomatic phase [4]. Therefore, we suggest that this clade be considered a new Variant of Interest (VOI), with the proposed lineage name P.1.2. Origin of this lineage was estimated to have happened around late January (Figure 4).

Figure 3. Genotype table (center) and Maximum Likelihood tree (left) of the new proposed lineage P.1.2 (highlighted in blue). Position of mutations are indicated by column names and base substitution is signalized by color change from gray. Bootstrap value is indicated on the tree.
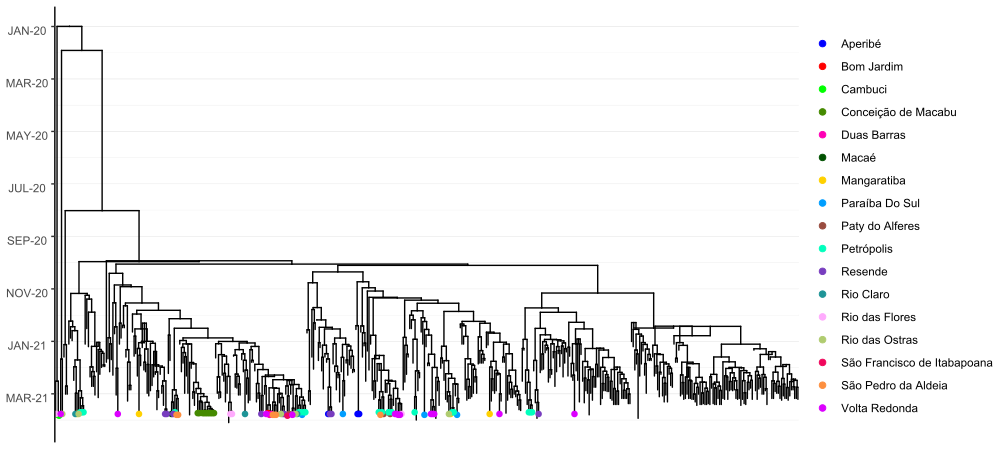
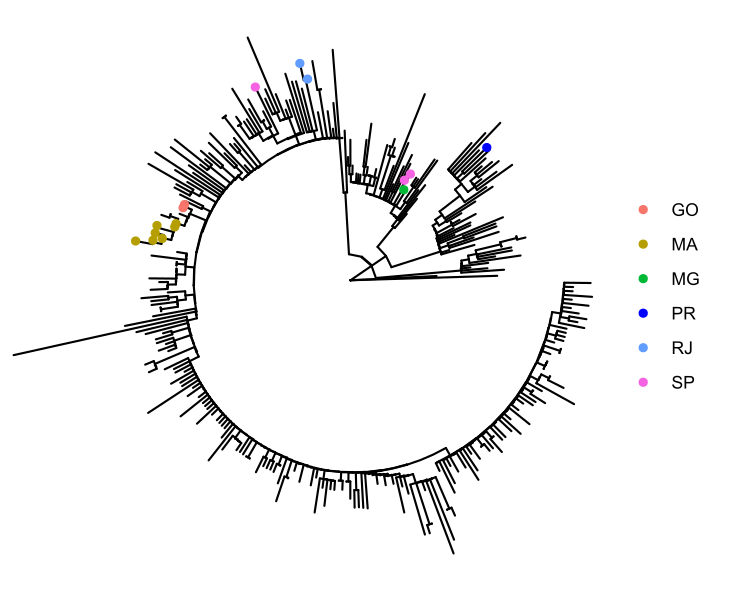
Figure 4. Time tree of the newly sequenced samples from Rio de Janeiro, Brazil, indicated by the points colored according to locality.
In addition to P.1 and P.2 lineages, we also found the B.1.1.7 lineage in three samples from the northwestern and southern regions of the state. These genomes harbored the E484K and N501Y mutations in the Spike protein, associated with the escape of the immunological system [5,6]. The B.1.1.7 lineage was first identified in the state in a sample from January, 1st, 2021 and its frequency has remained low (total n=8).
Conclusion
The identification of the quick replacement of P.2 by P.1 lineage through the sequencing of samples, reinforces the importance of continuosly genomic surveillance in the state of Rio de Janeiro aiming to monitor and prevent the strains dispersion. The adoption of new restrictive measures by the state and municipalities must consider the spatio-temporal distribution of the strains and the presence of mutations associated with the escape of the adaptive immune response.
Methods
The 90 analyzed genomes were collected between the dates March 24, 2021 and March 28, 2021 from 17 municipalities in the state of Rio de Janeiro/Brazil. Samples from patients with SARS-CoV-2 positive nasopharyngeal RT-PCR were collected at the Noel Nutels Central Laboratory (LACEN-RJ). The study was approved by the Ethics Committee (30161620.0.1001.5257 and 34025020.0.0000.5257). Patients were aged between 11 and 78 years old, being 50% men and 50% women. Extraction of the genetic material was performed at the Molecular Virology Laboratory (LVM-UFRJ) with QIAamp or MagMAX Viral / Pathogen Nucleic Acid Isolation kits and KingFisher automatic platform. Annealing of cDNA was conducted with 8,5 µl of viral RNA extracted from each sample. Libraries were constructed at the DFA/LNCC Genomics Unit with Illumina COVIDSeq Test (Illumina), according to the manufacturer’s protocol. Purification was then conducted using 5 µl of each library combined and the TapeStation (Agilent) system was used for quality control. We employed the MiSeq Reagent Kit v3 (600-cycle) to generate reads of 2x150 bp with a MiSeq sequencer. Sequence analysis, consensus building and variant calling were performed with DRAGEN COVID Lineage v3.5.1. Pango lineages were attributed to the newly assembled genomes using the PangoLEARN database v2021-04-14. They were then aligned with MAFFT 7.475 [7] to P.1 sequences and to sequences from Rio de Janeiro state regardless of lineage, all available in the GISAID database. The resulting alignment was then used to construct the maximum likelihood tree with IqTree2 [8] and the substitution model selected with the built-in algorithm ModelFinder [9]. Bootstrap support was calculated with 10,000 tree replicates. We also inferred the divergence dates of the new sequences using a bayesian approach with BEAST 1.10.4 [9,10]. Divergence dates were estimated by tip-dating and employing the strict clock model, the coalescent exponential growth tree prior and the GTR substitution model. Bioinformatic and phylogenetic analyses were performed using the computational infrastructure of the Bioinformatics Laboratory (LABINFO-LNCC).
Acknowledgments
We would like to thank all the authors and administrators of the GISAID database, which allowed this study of genomic epidemiology to be conducted properly. A full list acknowledging the authors publishing data used in this study can be found in the following file: gisaid_hcov-19_acknowledgement_table_2021_04_21_Post_Virological_02.pdf (226.3 KB)
This work was developed in the frameworks of Corona-ômica-RJ (FAPERJ = E-26/210.179/2020). A.T.R.V. is supported by CNPq (303170/2017-4) and FAPERJ (E-26/202.903/20); A.T. by FAPERJ E-26/010.002434/2019 and E-26/210.178/2020 R.S.F.J is a recipient of a graduate fellowship from CNPq, A.P.L is granted a post-doctoral scholarship (DTI-A) from CNPq.
We acknowledge the support from the Rede Corona-ômica BR MCTI/FINEP affiliated to RedeVírus/MCTI (FINEP 01.20.0029.000462/20, CNPq 404096/2020-4).
References
- Candido, D. S. et al. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science 369, 1255–1260 (2020).
- Voloch, C. M. et al. Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. J. Virol. (2021) doi:10.1128/JVI.00119-21
- Lamarca, A. P. et al. Genomic surveillance of SARS-CoV-2 tracks early interstate transmission of P.1 lineage and diversification within P.2 clade in Brazil. bioRxiv (2021) doi:10.1101/2021.03.21.21253418
- Gupta, S. et al. D155Y Substitution of SARS-CoV-2 ORF3a Weakens Binding with Caveolin-1: An in silico Study. bioRxiv (2021) doi:10.1101/2021.03.26.437194
- Collier, D. A. et al. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature (2021) doi:10.1038/s41586-021-03412-7
- Singh, J. et al. Structure-Function Analyses of New SARS-CoV-2 Variants B.1.1.7, B.1.351 and B.1.1.28.1: Clinical, Diagnostic, Therapeutic and Public Health Implications. Viruses 13, (2021).
- Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
- Minh, B. Q. et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 37, 1530–1534 (2020).
- Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
- Drummond, A. J. & Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007).