Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: preliminary findings
Nuno R. Faria1,2,3, Ingra Morales Claro3,4, Darlan Candido2,3, Lucas A. Moyses Franco3,4, Pamela S. Andrade3,4, Thais M. Coletti3,4, Camila A. M. Silva3,4, Flavia C. Sales3,4, Erika R. Manuli3,4, Renato S. Aguiar5, Nelson Gaburo6, Cecília da C. Camilo7, Nelson A. Fraiji8, Myuki A. Esashika Crispim8, Maria do Perpétuo S. S. Carvalho8, Andrew Rambaut9, Nick Loman10, Oliver G. Pybus2, Ester C. Sabino3,4, on behalf of CADDE Genomic Network11
- MRC Centre for Global Infectious Disease Analysis, J-IDEA, Imperial College London, London, United Kingdom.
- Department of Zoology, University of Oxford, Oxford, United Kingdom.
- Institute of Tropical Medicine, University of São Paulo, São Paulo, Brazil.
- Department of Infectious Disease, School of Medicine, University of São Paulo, São Paulo, Brazil.
- Departamento de Genética, Ecologia e Evolução, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil.
- DB Diagnósticos do Brasil, São Paulo, Brazil.
- CDL Laboratório Santos e Vidal Ltda., Manaus, Brazil.
- HEMOAM, Fundação de Hematologia e Hemoterapia do Amazonas, Manaus, Brazil.
- Institute of Evolutionary Biology, University of Edinburgh, Edinburgh, UK.
- Institute for Microbiology and Infection, University of Birmingham, Birmingham, UK.
- https://www.caddecentre.org/
Summary
We have detected a new variant circulating in December in Manaus, Amazonas state, north Brazil, where very high attack rates have been estimated previously. The new lineage, named P.1 (descendent of B.1.1.28), contains a unique constellation of lineage defining mutations, including several mutations of known biological importance such as E484K, K417T, and N501Y. Importantly, the P.1 lineage was identified in 42% (13 out of 31) RT-PCR positive samples collected between 15 to 23 December, but it was absent in 26 publicly available genome surveillance samples collected in Manaus between March to November 2020. These findings indicate local transmission and possibly recent increase in the frequency of a new lineage from the Amazon region. The higher diversity and the earlier sampling dates of P.1. in Manaus corroborates the travel info of recently detected cases in Japan, suggesting the direction of travel was Manaus to Japan. The recent emergence of variants with multiple shared mutations in spike raises concern about convergent evolution to a new phenotype, potentially associated with an increase in transmissibility or propensity for re-infection of individuals.
Context
Manaus, the largest city in the Amazon region, has been severely hit by the COVID-19 pandemic, with an estimated SARS-CoV-2 attack rate of three-quarters by October 2020 (Buss et al. 2020). Despite this, there is currently limited available genomic data from the region. Until 11 January 2021, existing genomes from Manaus were collected in March-April (1 reported in Valdinete et al. 2020, 6 from Candido et al. 2020; of these, 2 genomes belonged to the A2 lineage, 3 from B.1 and 2 from B.1.1.33). Since mid-December 2020 there has been a rapid increase in the number of cases and hospitalisations in Manaus (FVS/AM 2021; accessed 11 January 2021). Therefore, SARS-CoV-2 genome sequencing from more recent cases could help to shed light on the nature and diversity of lineages currently circulating in Manaus.
Study
In an attempt to investigate recently-circulating lineages in Manaus, we sequenced 37 samples collected from patients seeking private-healthcare PCR screening between 15 to 23 December 2020. All samples were PCR-confirmed and sequences were generated using the ARTIC v3 pipeline (Quick and Loman, 2020) using the MinION sequencing platform (Oxford Nanopore Technologies, ONT, UK). Negative controls for the sequencing libraries were clean. Consensus sequences were generated by the ARTIC pipeline 1.1.3 using nanopolish 0.13.2, followed by maximum likelihood phylogenetic analysis (Minh et al. 2020).
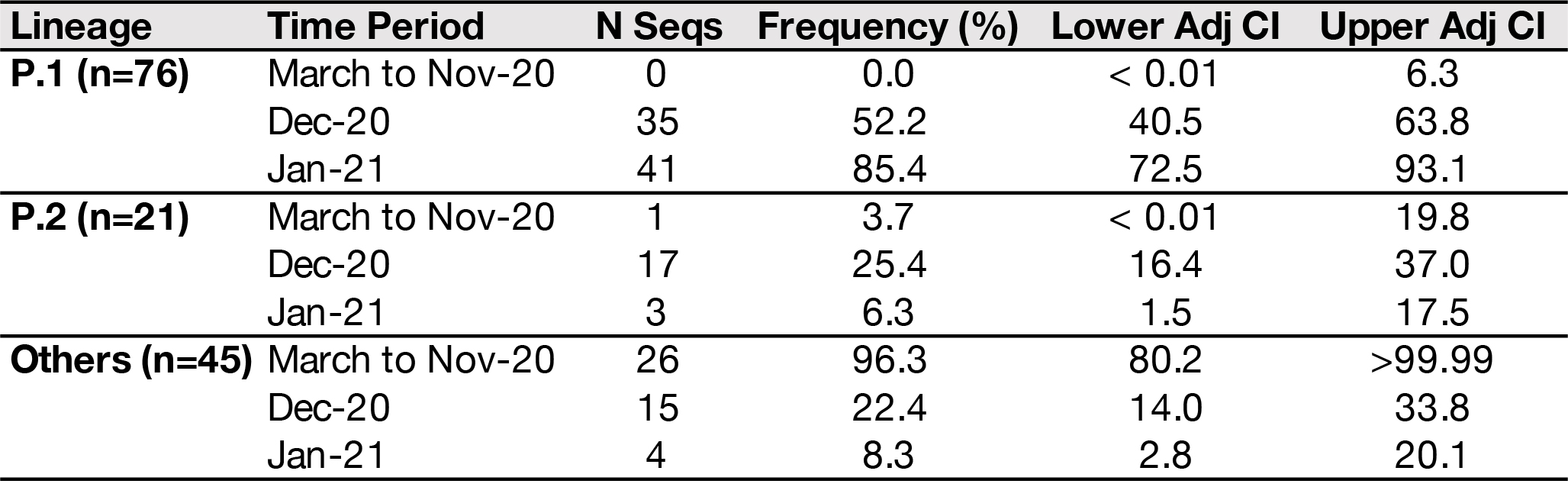
Of the 37 samples received, we generated 31 genomes with coverage >75% (mean 20x coverage of 88%, range: 62-97%). Genome sequences were classified using the latest pangolin version (2021-01-06, Rambaut et al. 2020; http://pangolin.cog-uk.io/). The lineage distribution for the newly-generated December genomes from Manaus was as follows: 65% B.1.1.28 lineage, 16% B.1.161, 11% B.1.1.33, 5% B.1.149, and 3% B.1.150. An alignment was generated with all available B.1.1.28 sequences from GISAID (https://www.gisaid.org/) downloaded on the 10 Jan 2021 (n=882) and a maximum likelihood tree was estimated for a total of 906 genomes. The 24 new B.1.1.28 sequences from Manaus (download here) fell on several distinct branches of the B.1.1.28 global phylogeny, suggesting multiple introductions of this lineage into Manaus (Fig. 1A), in line with previous observations using genomic data from March to November 2020 (Naveca et al. 2020).
However, we also detected a previously undiscovered novel cluster of B.1.1.28 genomes from Manaus containing a unique genetic signature (Fig. 1B). This new cluster, hereby named P.1 lineage, comprises 42% (13 out of 31) of the genomes from Manaus in mid/late-December and contains several mutations of potential biological significance. Here we provide a preliminary phylogenetic description and explain the nomenclature designation for this recently-detected lineage.
Lineage-defining mutations of the novel SARS-CoV-2 P.1 lineage
The new P.1 lineage carries 17 unique amino acid changes, 3 deletions, and 4 synonymous mutations, and one 4nt insertion compared to the most closely related available non-P.1 sequence (EPI_ISL_722052), which lies at the base of the long branch immediately ancestral to P.1 (Fig. 1B). The P.1 lineage meets the criteria for new lineage designation on the basis that it is phylogenetically and genetically distinct from ancestral viruses, associated with rapid spread in a new area, and carries a constellation of mutations that may have functional and/or phenotypic relevance (Rambaut et al. 2020). Although it is a descendent of B.1.1.28, it cannot be given the designation B.1.1.28.1, because lineage names cannot exceed three sublevels in the current classification system. Therefore, according to the dynamic nomenclature guidelines it is given the next available top-level designation, which is P.1 (Rambaut et al. 2020; http://pangolin.cog-uk.io/).
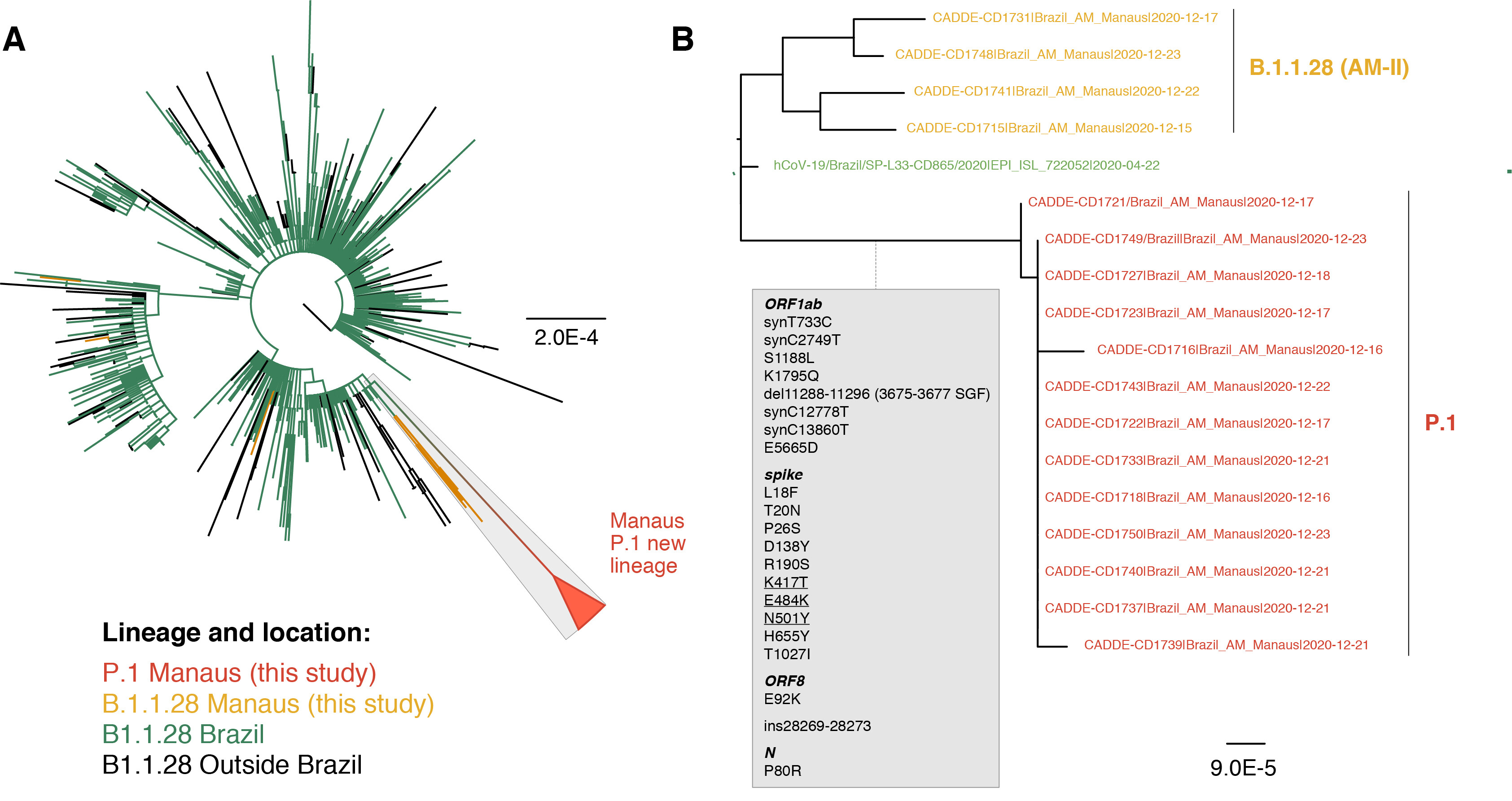
Figure 1 | Phylogenetic tree of the B.1.1.28 lineage rooted by its earliest genome (2020-03-05). A. Sequences generated in this study are highlighted in orange (n=18) and red (n=13, P.1. lineage). B. Phylogenetic tree highlighting the P.1 lineage containing the Manaus sequences and closely related sequences, including those from subclade AM-II. Inset in grey: the unique set of mutations from the P.1 lineage in comparison with its nearest sequence (EPI_ISL_722052). The scale of the phylogenetic branches is given as substitutions per nucleotide site.
Frequency of spike mutations E484K and N501Y in Brazil
We measured the frequency of two spike mutations of special interest among B.1.1.28 and its P.1. descendent lineage. The E484K mutation occurs in the receptor-binding domain (RBD) that the virus uses to bind to the human ACE2 receptor and has been associated with escape from neutralizing antibodies (Greaney et al. 2020). This variant now circulates throughout Brazil (Voloch et al. 2020; Naveca et al. 2020) and has been detected in a case of reinfection in Salvador, Bahia state (Nonaka et al. 2020). The frequency of the E484K among within B.1.1.28 lineage was 13% (n=100/750 genomes), while the frequency of E484K in the P.1 lineage was 100% (n=6/6 genomes with information at the position of interest). The N501Y mutation also occurs in the virus’ RBD. This mutation is associated with increased binding specificity and faster-growing lineages. This mutation is present in the P.1 lineage but has not been detected in Brazil, except in the two cases from a distinct B.1.1.7 lineage (Claro et al. 2020) and a single B.1 sequence from northeast Brazil (Paiva et al. 2020). The frequency of N501Y in the P.1 lineage was 100% (n=7/7 genomes with information at position of interest).
Convergent mutations shared between P1, B.1.1.7 and B.1.351 lineages
The newly described P.1. lineage from Manaus and the B.1.1.7 first described in the United Kingdom (Rambaut et al. 2020; Cov-Lineages) share the spike N501Y mutation and a deletion in ORF1b (del11288-11296 (3675-3677 SGF).
The P.1. lineage and the B.1.351 (also known as 501Y.V2) lineage described in South Africa (Tegally et al. 2020; Cov-Lineages) share three mutation positions in common in the spike protein (K417N/T, E484K, N501Y). Both the P.1 and the B.1.351 lineage also has the orf1b deletion del11288-11296 (3675-3677 SGF).
The set of mutations/deletions shared between P.1, B.1.1.7, and the B.1.351 lineages appear to have arisen entirely independently. Further, both mutations shared between P1 and B.1.351 seem to be associated with a rapid increase in cases in locations where previous attack rates are thought to be very high. Therefore it is essential to rapidly investigate whether there is an increased rate of re-infection in previously exposed individuals.
Limitations and future steps
These results should be considered with caution as the sample size is limited. Additional genome sequencing from the region is critically needed to investigate the frequency of the new variant over time, estimate its date of emergence, and infer population growth rates. Additional sequencing will also help to monitor local transmission of another epidemiologically relevant lineage, lineage B.1.1.7, which was discovered in the UK in early December 2020 (Rambaut et al. 2020) and has also been detected in two cases in Brazil 10 days later (Claro et al. 2020).
Data sharing
Genome data generated from Manaus has been deposited in GISAID with IDs EPI_ISL_804814 to EPI_ISL_804844. B.1.1.28 and P1 sequences can also be downloaded from CADDE center dedicated GitHub page. Genome sequences were obtained on 10 January 2021. Findings were shared with representatives from the World Health Organization, Pan American Health Organization, and Ministry of Health Colombia, Secretary of Health Amazonas, and FioCruz Manaus on 11 January 2021, during the elaboration of this report.
Acknowledgments
A full list acknowledging those involved in the diagnostics and generation of new sequences as part of the CADDE-Genomic-Network can be found in Table S1. We thank the administrators of the GISAID database for supporting rapid and transparent sharing of genomic data during the COVID-19 pandemic and to all our colleagues sharing data on GISAID. We also thank Dr. Felipe Naveca for sharing information ahead of publication. We thank Koen Deforche for identifying a shared mutation of interest within minutes of the original release of our report. A full list acknowledging the authors submitting B1.1.28 genome sequence data used in this study can be found in Table S2. Both supplementary tables can be found in GitHub - CADDE-CENTRE/Novel-SARS-CoV-2-P1-Lineage-in-Brazil.
Funding
This research was supported by a Medical Research Council-São Paulo Research Foundation (FAPESP) CADDE partnership award (MR/S0195/1 and FAPESP 18/14389-0) (CADDE Center). FAPESP further supports I.M.C. (2018/17176-8 and 2019/12000-1), J.G.J. (2018/17176-8 and 2019/12000-1, 18/14389-0), F.C.S.S. (2018/25468-9). N.R.F. is supported by a Wellcome Trust and Royal Society Sir Henry Dale Fellowship (204311/Z/16/Z). D.S.C. is supported by the Clarendon Fund and by the Department of Zoology, University of Oxford. O.G.P. is supported by the Oxford Martin School. This work received funding from the U.K. Medical Research Council under a concordat with the U.K. Department for International Development. N.J.L., and A.R. acknowledge the support of the Wellcome Trust (Collaborators Award 206298/Z/17/Z ARTIC network). A.R. is supported by the European Research Council (grant agreement no. 725422, ReservoirDOCS). We acknowledge support from the Rede Corona-ômica BR MCTI/FINEP affiliated to RedeVírus/MCTI (FINEP 01.20.0029.000462/20, CNPq 404096/2020-4). We additionally acknowledge support from Community Jameel and the NIHR Health Protection Research Unit in Modelling Methodology.
Contact information
Prof. Nuno R. Faria (Imperial College London, University of Oxford, Instituto Medicina Tropical, University of São Paulo): Email: [email protected]
Prof. Ester C. Sabino (Instituto de Medicina Tropical da Faculdade de Medicina, University of São Paulo): Email: [email protected]