Detection of non-B.1.1.7 Spike ∆69/70 sequences (B.1.375) in the United States
We currently caution against using TaqPath S dropout as a proxy for B.1.1.7 in the US
Gage Moreno1, Katarina Braun2, Brendan B. Larsen3, Tara Alpert4, Michael Worobey3, Nathan Grubaugh4, Thomas Friedrich2, David O’Connor1, Joseph Fauver4, and Anderson Brito4
- Department of Pathology and Laboratory Medicine, University of Wisconsin - Madison
- Department of Pathobiological Sciences, University of Wisconsin-Madison
- Department of Ecology and Evolutionary Biology, University of Arizona
- Department of Epidemiology of Microbial Diseases, Yale School of Public Health
Summary
Multiple SARS-CoV-2 variants are circulating globally, however, phenotypic differences, if any, have yet to be described for the majority of these variants. Routine surveillance sequencing of SARS-CoV-2 is critical for identifying and tracking new mutations. Most notably, two recently emerged lineages, termed B.1.1.7 (also known as 20B/501Y.V1, and Variant of Concern (VOC) 202012/01), and B.1.351 (20C/501Y.V2), have become “variants of concern.”
B.1.1.7 lineage (20B/501Y.V1) has 17 amino acid substitutions relative to the initial SARS-CoV-2 genome sequence. In particular, investigations have focused on mutations in the Spike attachment protein, including deletions of amino acids 69 and 70 (S ∆69/70), N501Y, and P681H, as well as ORF8 Q27stop. The B.1.1.7 lineage is thought to have increased transmissibility based on recent epidemiological and phylogenetic data1.
The Spike ∆69/70 deletion prevents the oligonucleotide probe used in the commercial Applied Biosystems TaqPath COVID-19 assay (ThermoFisher) from binding its target sequence, leading to what has been termed “S gene dropout” or “S gene target failure,” that is, a lack of signal from the S gene amplicon, together with successful amplification of other two SARS-CoV-2 gene targets2. It was quickly recognized that this S gene target failure could be used as a proxy for B.1.1.7 lineage viruses, at least when B.1.1.7 had already reached some prevalence in the community3. Because the S ∆69/70 deletion occurs in other genetic backgrounds in addition to B.1.1.7, sequencing is necessary to confirm the genetic lineage of a virus with S gene target failure, particularly in regions where B.1.1.7 is not already known to circulate. However, viruses displaying S gene target failure in the U.S. should be prioritized for follow-up sequencing as the proportion of these failures attributable to B.1.1.7 viruses appears to vary geographically4.
Study
A recent post by Brendan Larsen and Michael Worobey details a novel lineage (now named B.1.375, in a recent update of pangolin5) circulating in the United States that contains S ∆69/70, but no other B.1.1.7 mutations. Wisconsin and Connecticut have found similar S ∆69/70 non-B.1.1.7 sequences. The Grubaugh lab has primarily identified S ∆69/70 sequences by targeted sequencing of S-gene dropouts, whereas the O’Connor and Friedrich labs have focused on broad surveillance sequencing in Dane County, Wisconsin. Currently, despite their similarity, these sequences were initially assigned to a number of discrete Pangolin lineages. The Pangolin team has been made aware of this and subsequently retrained PangoLEARN to name this lineage B.1.375.
Evolutionary history and mutations associated with B.1.375
Here, building off of the Worobey lab’s previous post, we present an additional 27 SARS-CoV-2 genomes released as of the early hours of January 11th, 2021 which contain the ∆69/70 and are not B.1.1.7 lineage. This pattern was revealed in a maximum likelihood phylogenetic tree inferred using augur6 (see nextrain build here), highlighting the evolutionary history of these variants with respect to viruses from other lineages (Figure 1). We obtained this tree by subsampling the set of genomes submitted to GISAID up to January 11th, from lineages B.1.1.7, B.1.351, and B.1.346, as well as contextual genomes from the US, and global genomes from Oceania, (South) Africa, Europe and Asia. Additionally, the dataset was enriched with the latest US genomes with S ∆69/70 (see acknowledgements, Table 1).
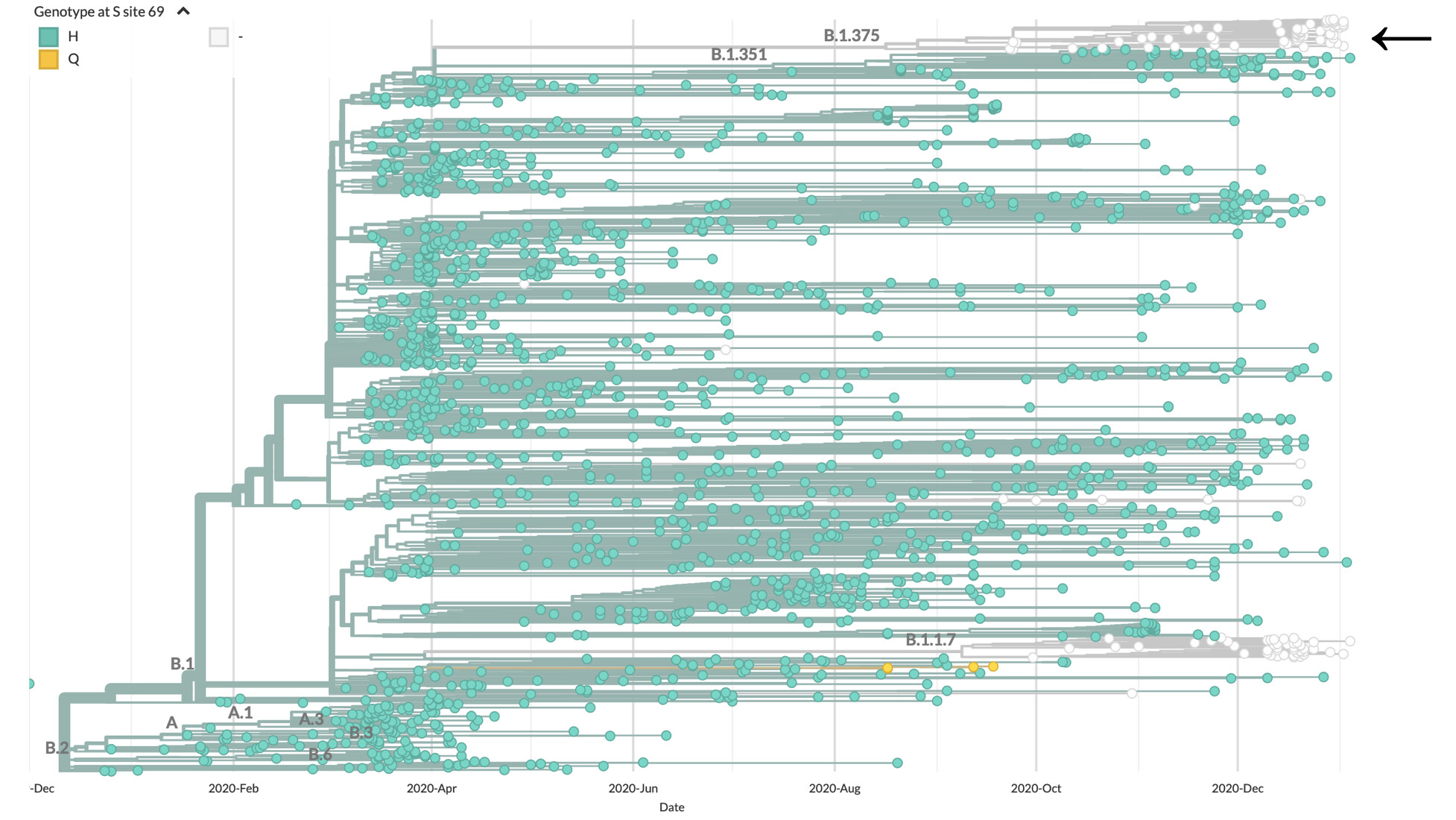
Figure 1. Evolutionary relationship among variants from the new lineage B.1.375 detected in the US (white circles, see arrow at the top), and other variants circulating nationally and internationally. The ‘UK variant’ (lineage B.1.1.7) is shown at the bottom, also as white circles, indicating the presence of the deletion S ∆69/70, also observed in the unrelated lineage we show in this report (interactive tree available here).
This analysis revealed a monophyletic cluster of genomes (Figure 2A) defined by 5 amino acid substitutions: ORF1a T1828A, ORF1b E1264D, ORF3a T151I, S ∆69/70, M I48V (Table 1). This lineage, which is observed in at least 12 US states in all regions of the country (Figure 2B), has been circulating locally, within the United States, since at least mid-September 2020 when it was first detected.
Table 1. List of nucleotide and amino acid substitutions that are shared among the B.1.375 lineage viruses.
| Gene | Amino acid substitution | Nucleotide substitution |
|---|---|---|
| ORF1a | T1828A | A5747G |
| ORF1b | E1264D | G17259T |
| ORF3a | T151I | C25884T |
| Spike | ∆69/70 | T21765-, A21766-, C21767-, A21768-, T21769-, G21770- |
| Membrane | I48V | A26664G |
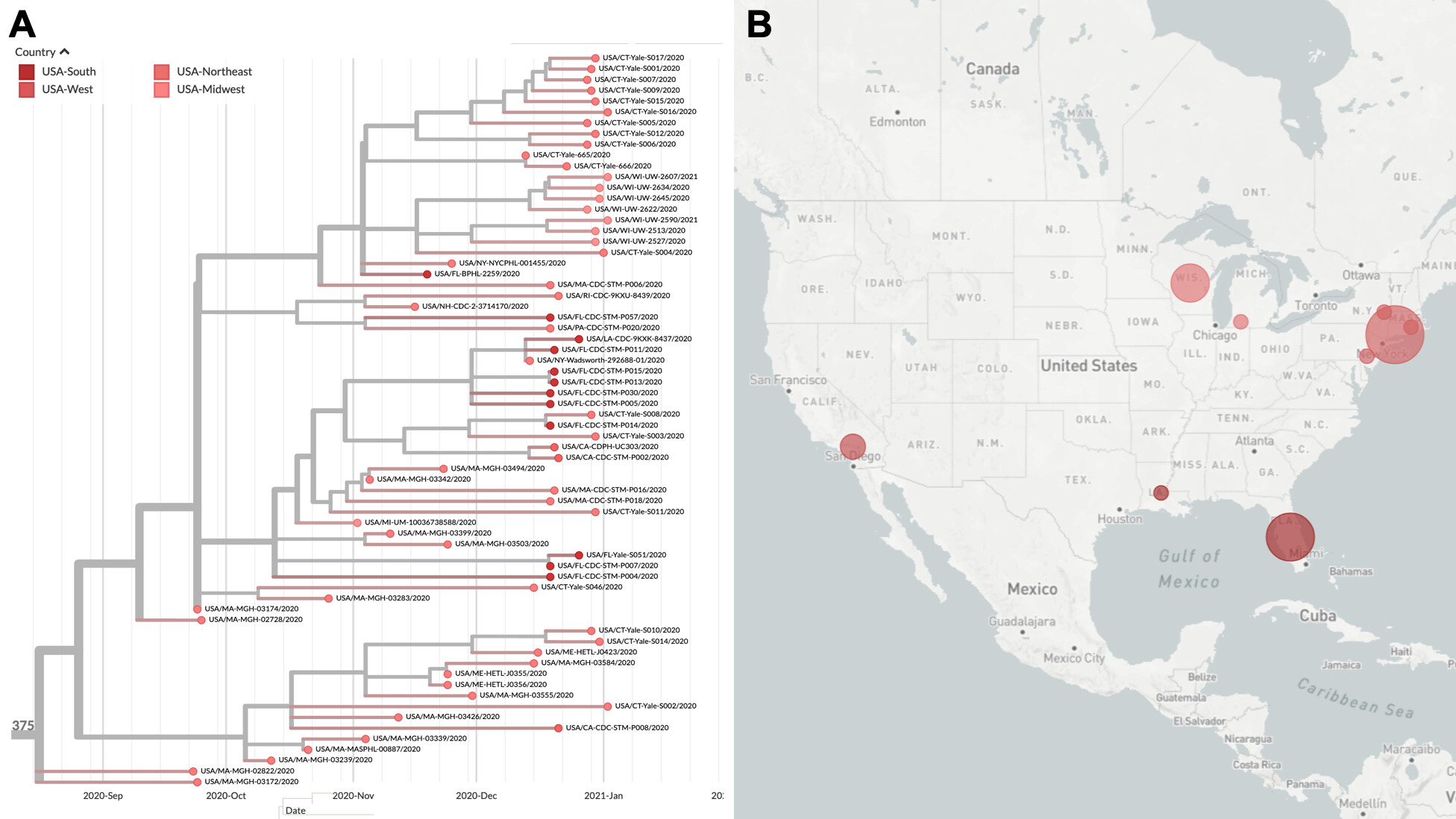
Figure 2. B.1.375 lineage of US variants. A) Clade highlighting new genomes sequenced by distinct groups in the US since September, and found in all regions of the country. All these genomes express the deletion S ∆69/70, but are unrelated to B.1.1.7. B) Geographic distribution of variants belonging to this US lineage (interactive tree available here).
Caution against using TaqPath S dropout as a proxy for B.1.1.7
The detection of viruses from unrelated lineages displaying S ∆69/70, such as the new one here reported (B.1.375), emphasizes that while using the TaqPath S-dropout approach as a proxy for B.1.1.7-lineage detection in the UK appears to work well, we currently caution against using this approach in the United States as there are clearly non-B.1.1.7 sequences containing the ∆69/70 that are more common than B.1.1.7. Overall, B.1.375 remains at very low levels compared to the total number of genomes sequenced from the United States. However, in Wisconsin where sequencing was non-targeted surveillance, 4% of all sequences contained S ∆69/70 from December 29, 2020 to January 4, 2020. However, the relatively high prevalence of these sequences in Wisconsin could be due to cases identified as part of a local cluster. Continued surveillance of this cluster in Wisconsin is necessary to determine if this clade is outpacing other circulating variants.
Limitations and future steps
We want to emphasize that there is no data to suggest that variants from B.1.375 are more transmissible or cause more severe illness, or increased risk of death. The tracking the spread of these variants, and the potential for new variants to arise, requires enhanced genomic surveillance.
The Grubaugh lab has already begun work to develop a RT-qPCR-based screening strategy to differentiate between ∆69/70 B.1.1.7 and ∆69/70 B.1.375. They currently have a protocol that recreates the “S gene target failure” signature - essentially a TaqPath assay “hack” which can be found here. They are working on expanding this as a more specific screening tool for B.1.1.7 by detecting two S dropouts (one at ∆69/70 and one further downstream at ∆144) that are not currently found in other virus lineages (see dedicated virological post).
The Worobey group has also developed high-stringency PCR assays to detect B.1.1.7 from a variety of SARS-CoV-2 sample types. They have designed pairs of primers, one pair that amplifies a product in the vicinity of the S 69-70 deletion, another in the vicinity of the S 144 deletion, with either the forward or reverse primer in each pair binding to the B.1.1.7 spike gene in the deletion zone. Hence these primers encounter “extra” nucleotides on spike genes of viruses lacking these deletions and fail to bind effectively. Positive control oligonucleotides with and without the deletions (B.1.1.7-like and non-B.1.1.7-like, respectively) as well as patient samples indicate high sensitivity and specificity, with sharp bands appearing in gels when the deletions are present, and no bands when the deletions are absent. They are currently screening wastewater samples with this approach, but it is amenable to patient samples too, including pools of samples that may contain rare B.1.1.7 cases. Details of the protocols will be shared on virological.org shortly.
Acknowledgements
We gratefully acknowledge the laboratories and researchers who made these SARS-CoV-2 genomes available on GISAID (Table 2). We also thank Áine Toole, Emily Scher and the Rambaut group for quickly training pangoLEARN with a new model to assign the new lineage B.1.375 5.
Contact information
Anderson F. Brito (Yale School of Public Health, Department of Epidemiology of Microbial Diseases): Email: [email protected]
Joseph Fauver (Yale School of Public Health, Department of Epidemiology of Microbial Diseases): Email: [email protected]
Gage Moreno (Department of Pathology and Laboratory Medicine, University of Wisconsin - Madison): Email: [email protected]
Table 2. List of genomes from the new lineage B.1.375, which also show the deletion ∆69/70 (unrelated to B.1.1.7) used in this preliminary study. We thank the authors of all 1,699 genomes used in Figure 1, which are listed in acknowledgements.pdf (242.3 KB), which outlines the author contributions.
| Author | Number of genomes | Strains |
|---|---|---|
| Lemieux et al A | 14 | USA/MA-MGH-03172/2020,USA/MA-MGH-02822/2020,USA/MA-MGH-03239/2020,USA/MA-MGH-03339/2020,USA/MA-MGH-03426/2020,USA/MA-MGH-03555/2020,USA/MA-MGH-03584/2020,USA/MA-MGH-02728/2020,USA/MA-MGH-03174/2020,USA/MA-MGH-03283/2020,USA/MA-MGH-03503/2020,USA/MA-MGH-03399/2020,USA/MA-MGH-03342/2020,USA/MA-MGH-03494/2020 |
| Andrew Lang et al | 1 | USA/MA-MASPHL-00887/2020 |
| Peter W. Cook et al | 15 | USA/CA-CDC-STM-P008/2020,USA/FL-CDC-STM-P004/2020,USA/FL-CDC-STM-P007/2020,USA/MA-CDC-STM-P018/2020,USA/MA-CDC-STM-P016/2020,USA/CA-CDC-STM-P002/2020,USA/FL-CDC-STM-P014/2020,USA/FL-CDC-STM-P005/2020,USA/FL-CDC-STM-P030/2020,USA/FL-CDC-STM-P013/2020,USA/FL-CDC-STM-P015/2020,USA/FL-CDC-STM-P011/2020,USA/PA-CDC-STM-P020/2020,USA/FL-CDC-STM-P057/2020,USA/MA-CDC-STM-P006/2020 |
| Fauver et al | 20 | USA/CT-Yale-S002/2020,USA/CT-Yale-S014/2020,USA/CT-Yale-S010/2020,USA/CT-Yale-S046/2020,USA/FL-Yale-S051/2020,USA/CT-Yale-S011/2020,USA/CT-Yale-S003/2020,USA/CT-Yale-S008/2020,USA/CT-Yale-S004/2020,USA/CT-Yale-666/2020,USA/CT-Yale-665/2020,USA/CT-Yale-S006/2020,USA/CT-Yale-S012/2020,USA/CT-Yale-S005/2020,USA/CT-Yale-S016/2020,USA/CT-Yale-S015/2020,USA/CT-Yale-S009/2020,USA/CT-Yale-S007/2020,USA/CT-Yale-S001/2020,USA/CT-Yale-S017/2020 |
| Matluk et al | 3 | USA/ME-HETL-J0356/2020,USA/ME-HETL-J0355/2020,USA/ME-HETL-J0423/2020 |
| Valesano et al | 1 | USA/MI-UM-10036738588/2020 |
| Charles Chiu et al | 1 | USA/CA-CDPH-UC303/2020 |
| Kirsten St. George et al | 1 | USA/NY-Wadsworth-292688-01/2020 |
| Ying Tao Yan Li Jing Zhang Krista Queen Anna Uehara Peter Cook Clinton R. Paden Haibin Wang Suxiang Tong et al | 2 | USA/LA-CDC-9KXK-8437/2020,USA/RI-CDC-9KXU-8439/2020 |
| Krista Queen et al | 1 | USA/NH-CDC-2-3714170/2020 |
| Sarah Schmedes et al | 1 | USA/FL-BPHL-2259/2020 |
| Jade Wang et al | 1 | USA/NY-NYCPHL-001455/2020 |
| Gage Moreno et al | 7 | USA/WI-UW-2527/2020,USA/WI-UW-2513/2020,USA/WI-UW-2590/2021,USA/WI-UW-2622/2020,USA/WI-UW-2645/2020,USA/WI-UW-2634/2020,USA/WI-UW-2607/2021 |
References
- Center for Disease Control. Emerging SARS-CoV- Variants. (2020). https://www.cdc.gov/coronavirus/2019-ncov/more/science-and-research/scientific-brief-emerging-variants.html
- Kidd, M. et al. S-variant SARS-CoV-2 is associated with significantly higher viral loads in samples tested by ThermoFisher TaqPath RT-QPCR. medRxiv 2020.12.24.20248834 (2020).
- Public Health England. Investigation of novel SARS-CoV-2 variant: Variant of Concern 202012/01. (2020). https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/947048/Technical_Briefing_VOC_SH_NJL2_SH2.pdf
- Nicole Washington, S. W., Kelly Schiabor Barrett, Elizabeth Cirulli, Alexandre Bolze, James Lu. Update on the Helix, Illumina surveillance program: B.1.1.7 variant of SARS-CoV-2, first identified in the UK, spreads further into the US. (2020). https://blog.helix.com/b117-variant-updated-data/
- Áine O’Toole, J. T. M., Emily Scher. Phylogenetic Assignment of Named Global Outbreak LINeages. (2020). GitHub - cov-lineages/pangolin: Software package for assigning SARS-CoV-2 genome sequences to global lineages.
- Hadfield, J. et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34, 4121-4123 (2018).