Background
The P.1 lineage of SARS-CoV-2, recognized as a Variant of Concern (VoC) by the US CDC and WHO 1, is characterized by 17 mutations including several known or suspected to have functional consequences. P.1 lineage viruses are associated with marked reductions in neutralizing antibodies from convalescent and vaccinated patients 2 and have been found in reinfection cases 3. This lineage is also reported to be more transmissible 4. Consequently, P.1 may pose significant risks to both individual and public health. P.1 is currently the least documented VoC in the United States, possibly a result of restricted travel from areas where the lineage is prevalent; however, an increasing number of states are now reporting P.1 cases 5.
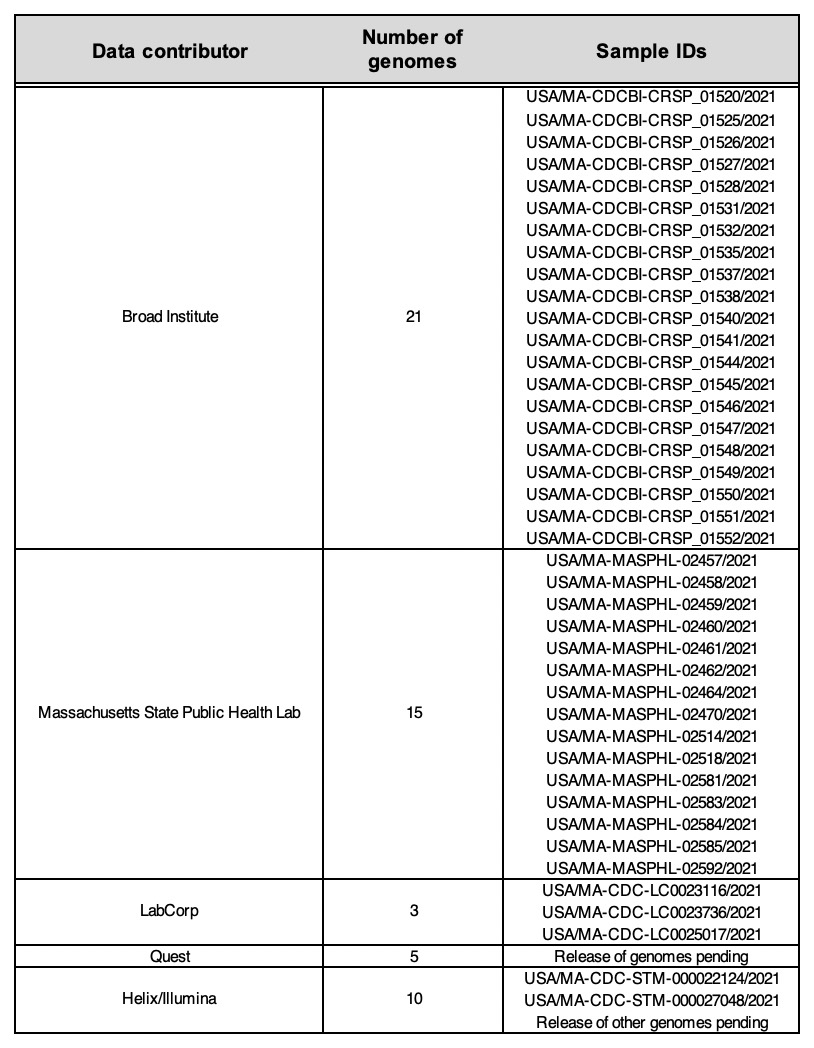
To support state public health investigations, we have been sequencing SARS-CoV-2 viral genomes from cases in Massachusetts (MA) and New England more broadly, with a recent focus on the Cape Cod area following a notable increase in cases and the presence of a putative cluster there. Several of these recent genomes were found to be P.1. In parallel, a combination of routine state-wide genomic surveillance (by the CDC Baseline Surveillance program’s contract labs), and focal sequencing around the suspected cluster (by the MA State Public Health Laboratory (MASPHL)), has identified additional P.1 cases in Cape Cod and across the state. Here we describe a dataset of 54 high-quality genomes of the P.1 VoC lineage from unique patient samples obtained through these combined efforts (see Table 1).
Results
All three global VoCs (B.1.1.7, B.1.351, and P.1) have now been detected in MA. However, unlike B.1.1.7 and B.1.351, P.1 has shown a rapid rise in frequency in the few weeks following its first detection on March 16th, 2021 (Figure 1). Phylogenetic analysis of these 54 P.1 genomes from MA, alongside four additional P.1 genomes from neighbouring states (two from Connecticut (CT) and one each from Maine (ME) and Rhode Island (RI)) and 895 other global P.1 sequences from GISAID, 6 suggests at least six introductions of P.1 into MA and at least eight introductions of P.1 into the New England region, likely from both international and domestic sources (Figure 2).

Figure 1. Detection of Variants of Concern in Massachusetts. A. Numbers of SARS-CoV-2 genomes from Massachusetts by week of collection since October 31st 2020. A. Counts represent all data available in GISAID as of April 1, 2021, plus 14 additional genomes from Massachusetts that will be released shortly. Genomes of the three global Variants of Concern are highlighted in colour. B. shows the same data with 20 Massachusetts P.1 genomes identified through focal testing around a suspected cluster of cases removed.
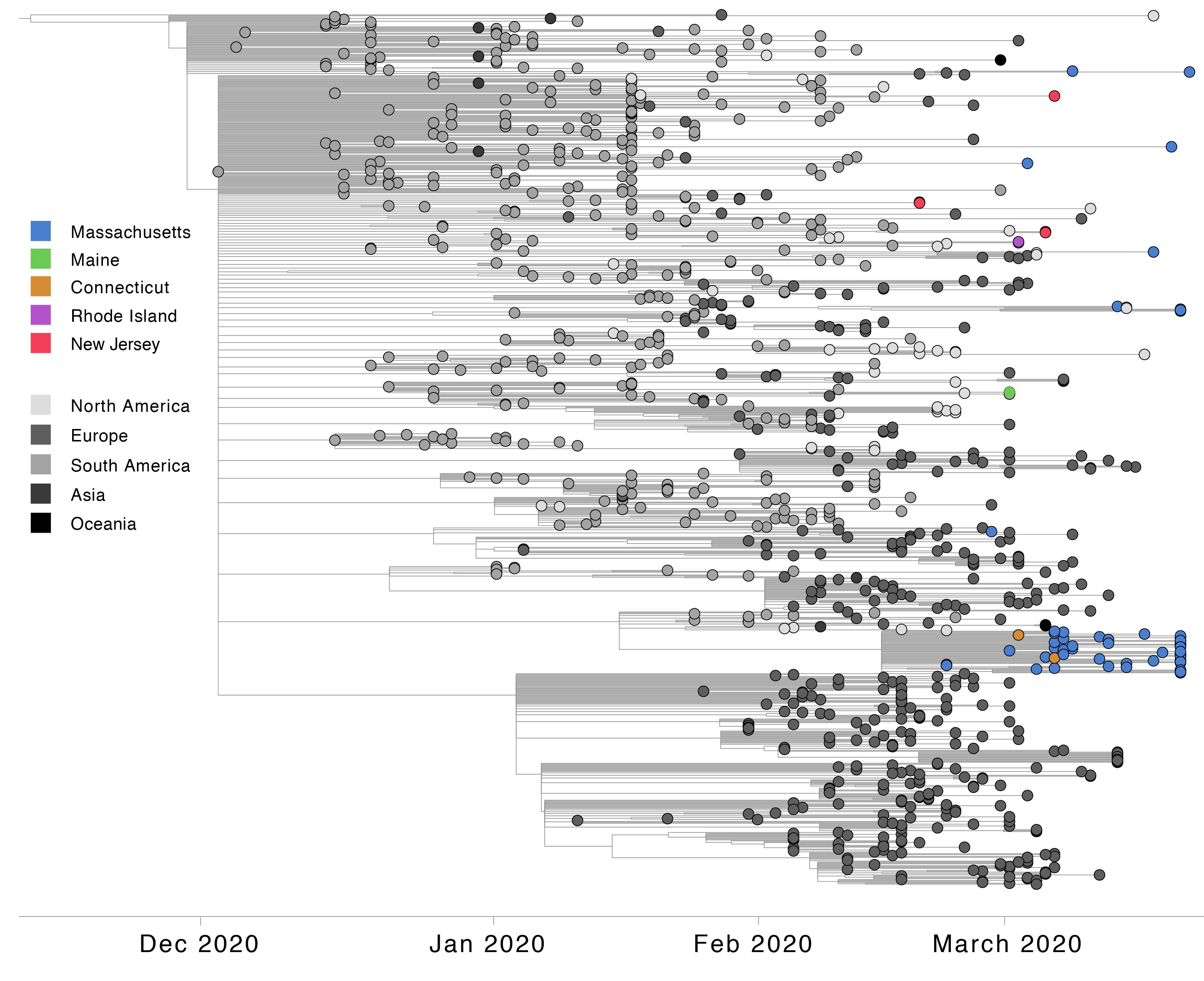
Figure 2. Time tree of 952 P.1 genomes, including 54 from Massachusetts and 898 selected from GISAID on Mar 30, 2021. Genomes from Massachusetts and other New England states are highlighted in color.
Of 54 MA P.1 genomes, 43 form a single, distinct monophyletic clade, suggesting they are part of a transmission cluster (Figure 3). Based on ancestral inference, this cluster likely originated from a single introduction from Brazil. The two P.1 genomes on GISAID from CT are identical to the predominant sequence of the MA cluster, suggesting that P.1 may have been introduced into CT from MA, although no epidemiological link has been identified. P.1 genomes in the MA cluster are highly similar (0-4 SNP differences). Of the 43 genomes in the cluster, 28 genomes, from samples collected in a 13 day period (3-21 March), are genetically identical, a pattern consistent with a potential superspreading event. These genomes are characterized by 3 amino acid altering mutations that uniquely distinguish this MA clade from P.1 genomes globally (ORF1a G2941S, ORF1b A1219S, and ORF3a Q57H). This clade also contains 10 amino acid altering mutations, including four in the Spike protein, segregating within the cluster.

Figure 3. Maximum likelihood tree of 43 genomes from Massachusetts and two genomes from Connecticut that form a closely related monophyletic cluster.
A second, smaller cluster of P.1 genomes contains four genomes from MA and two from Nebraska, from samples collected close in time to the MA samples. The direction of the link between the two sets cannot be reliably determined. The remaining five MA P.1 genomes likely represent four or five additional independent introductions into the state, given their substantial genetic distances from each other and the other MA genomes (4-17 SNP differences). Two of these genomes cluster with several genomes from Italy, suggesting a probable P.1 importation from Europe.
The single P.1 genomes available from Maine and Rhode Island are genetically distinct from others currently detected in New England, suggesting that they are further independent introductions into the region. Notably, these P.1 genomes cluster with genomes from Florida and Washington, respectively, suggesting domestic spread of the P.1 variant between New England and other US states.
Discussion
The detection of over 50 cases of the P.1 variant, a VoC that has been associated with increased transmissibility and reinfection, in less than a month in MA is concerning.
That many of these genomes appear to reflect multiple independent introductions of P.1 into New England, and transportation of this variant between several US states, points to likely widespread circulation of this VoC across the country. While our understanding of the increased transmissibility of the P.1 variant is still unfolding, the large cluster of identical and nearly identical genomes–a pattern of genetic diversity consistent with superspreading-- highlights the ongoing risk of rapid spread of SARS-CoV-2, and in particular for more transmissible lineages. The extent of community transmission of P.1 in Massachusetts and across the US is currently unknown, however, it is likely that the P.1 variant is already or will soon be circulating in communities and ongoing surveillance will be critical to understanding the trajectory and impact of the P.1 variant.
Methods
Viral sequencing was performed by the Massachusetts State Public Health Laboratory and by four CDC Baseline Surveillance contract labs: Quest, Labcorp, Helix/Illumina, and the Broad Institute. Sequencing at the Broad Institute was performed with approval from the MIT Institutional Review Board (protocol #1612793224). Most laboratories used multiplexed amplicon-based sequencing methods based on the ARTIC V3 7 primer set followed by Illumina sequencing (Labcorp utilized PacBio sequencing with CLC Genomics analyses). Contract genomes were provided to MA DPH by the CDC prior to public data release. Phylogenetic analyses were conducted at the Broad Institute utilizing the Nextstrain pipeline 8 adapted to a cloud compute workflow (link).
Data availability
An interactive phylogeny including this cluster is available at the following URL: https://auspice.broadinstitute.org/sars-cov-2/ma-cluster/20210322?f_Nextstrain_clade=20J/501Y.V3&f_region=North%20America&label=clade:20J/501Y.V3&m=div
Genomes from the Broad Institute are available on NCBI Genbank and SRA under BioProject PRJNA715749 and on GISAID. Genomes from MADPH and one from Helix/Illumina are available on GISAID. Remaining genomes are expected to be released by the CDC shortly.
Table 1. Data contributors to SARS-CoV-2 P.1 genomes from Massachusetts.
Acknowledgements
We thank all groups that contributed to the generation of these genomes from New England, including the Massachusetts Department of Public Health, the Rhode Island Department of Health, Quest Diagnostics, Labcorp, Helix/Illumina, and all individuals and organizations that submitted samples for sequencing. We gratefully acknowledge the authors from the originating laboratories and the submitting laboratories who generated and shared via GISAID genetic sequence data that was used in this analysis (Table 2), in particular Maine Health and Environmental Testing Laboratory, the Tewhey Lab at The Jackson Laboratory, Yale Clinical Virology Lab, and the Grubaugh Lab at Yale School of Public Health.
This work was supported by the Centers for Disease Control and Prevention’s Baseline Surveillance program (Broad Institute, Quest, Labcorp, Helix/Illumina), a Centers for Disease Control and Prevention Broad Agency Announcement (75D30120C09605 to Broad Institute), the National Institute of Allergy and Infectious Diseases (U19AI110818 to Broad Institute), and the Centers for Disease Control and Prevention cooperative agreement “Building and Enhancing Epidemiology, Laboratory and Health Information Systems Capacity in Massachusetts” grant (6 NU50CK000518 to MADPH).
Pardis C. Sabeti is a co-founder and shareholder of Sherlock Biosciences and is a non-executive board member and shareholder of Danaher Corporation.
Contributors
Project team and contributors includes, from MA Department of Public Health: Catherine Brown, Matthew Doucette, Timelia Fink, Glen Gallagher, Andrew Lang, Larry Madoff, Sandra Smole; RI Department of Health: Kristin Azevedo, Richard Huard, Ewa King, Adam Miller; Broad Genomics Platform: Mike DaSilva, Nick Fitzgerald, Zoe Mandese, Tamara Mason, Samantha McGovern, Maesha Ulcena, Gina Vicente, Brendan Blumenstiel, Matthew DeFelice, Matthew Lee, Jim Meldrim, Brian Granger, Katie Larkin, Sheila Dodge, Niall Lennon, Stacey Gabriel; Broad Data Sciences Platform: Sushma Chaluvadi, Christine Loreth, Anthony Philippakis, DSP Field Engineering Team; Massachusetts General Hospital: Jacob Lemieux, Bennett Shaw; Broad Viral Genomics Group: Gordon Adams, Matthew Bauer, Amber Carter, Kat DeRuff, Adrianne Gladden-Young, Lydia A. Krasilnikova, Kim Lagerborg, Bronwyn MacInnis, Erica Normandin, Danny Park, Leah Pearlman, Steve Reilly, Melissa Rudy, Pardis Sabeti, Stephen Schaffner, Katherine Siddle, Jillian Silbert, Chris Tomkins-Tinch
Contact
[email protected] @dannyjpark
[email protected] @bronwynmacinnis
References
-
Wang, P. et al. Increased Resistance of SARS-CoV-2 Variant P.1 to Antibody Neutralization. bioRxiv 2021.03.01.433466 (2021) doi:10.1101/2021.03.01.433466.
-
Coutinho, R. M. et al. Model-based estimation of transmissibility and reinfection of SARS-CoV-2 P.1 variant. bioRxiv (2021) doi:10.1101/2021.03.03.21252706.
-
Faria, N. R. et al. Genomics and epidemiology of a novel SARS-CoV-2 lineage in Manaus, Brazil. medRxiv (2021) doi:10.1101/2021.02.26.21252554.
-
CDC. Variant Proportions in the U.S. https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/variant-proportions.html (2021).
-
Tyson, J. R. et al. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. bioRxiv 2020.09.04.283077 (2020) doi:10.1101/2020.09.04.283077.
Table 2. GISAID author acknowledgements
gisaid_author_acknowledgement.tsv.zip (26.0 KB)