Jonas Fuchs1 and Anton Nekrutenko2, Ann-Kathrin Kohl1, Kristin Technau-Hafsi3, Daniel Hornuß4, Siegbert Rieg4, Lena Jaki1, Daniela Huzly1, Wolfgang Maier5, Björn Grüning5, Lisa Gutbrod1, Valeria Falcone1, Hartmut Hengel1, Marcus Panning1
1Institute of Virology, Freiburg University Medical Center, Freiburg, Germany
2The Pennsylvania State University, University Park, PA, USA
3Department of Dermatology and Venereology, Freiburg University Medical Center and Faculty of Medicine, Freiburg, Germany
4Department of Internal Medicine II, Division of Infectious Diseases, Freiburg University Medical Center and Faculty of Medicine, Freiburg, Germany
5Department of Computer Science, Albert-Ludwigs-University, Freiburg, Freiburg, Germany
Introduction
The Monkeypox virus (MPXV) is a rare zoonotic pathogen belonging to the Orthopox virus genus that includes vaccinia virus, ectromelia virus, camelpox virus, cowpox viruses and the devastating variola virus globally eradicated in 1980. MPXV has a wide host range, enabling it to maintain its natural reservoir in wild rodent species, and can be transmitted to humans causing varying disease severity with skin lesions. Typically, a MPXV rash starts 10 to 14 days after infection with small lesions that spread in a centrifugal pattern [1,2]. The first outbreak in humans was reported in 1970 in the Democratic Republic of Congo [3]. After that, human cases have been primarily reported from central and western Africa with only a few cases outside of the African continent [4].
Since May 2022, over 700 human cases in 27 non-endemic countries have been confirmed (accessed on 2022-02-06). Here, we report the MPXV genome sequences of two male patients that presented symptomatic in the last week of May at the Freiburg University Medical Center, Germany, after traveling back from the Grand Canary Island, Spain. Moreover, we were able to isolate infectious virus from skin swabs under biosafety level 3 conditions and generated the first genome sequence of a virus isolate of the 2022 MPXV cluster.
Descriptions of the methods
Virus was isolated from swabs of skin lesions on VeroE6 cells. The cytopathic effect was apparent four days after infection and grew radially indicating an efficient cell to cell spread and plaque formation. Six days after infection, DNA was isolated from viral supernatant with the Monarch® Nucleic Acid Purification Kits (NEB). DNA from skin lesion swabs was extracted with the DNA Blood mini Kit (QIAGEN). The extracted total DNA of patient samples and the virus isolate were used to generate paired-end libraries via the NEBNext® Ultra™ II DNA Library Prep Kit (NEB) and sequenced on an Illumina Miseq instrument (2x150 bp). The samples of the two patients were sequenced in independent runs. In cooperation with Galaxy Project, we developed a series of workflows for the detection of MPXV sequence variants. Briefly, raw reads were processed with fastp (v0.20.1) and mapped to the MT903343 reference genome using BWA-MEM (v0.7.17). Viral variants were called with lofreq (v2.1.5) and annotated with snpeff (v4.3.1). Consensus sequences were generated with bcftools (v.1.10) masking low covered regions (≥ 5x) and variant frequencies between 0.3 and 0.7 with Ns. We downloaded 114 MPXV full-length genomes from NCBI and included our own sequences and the sequences shared by our colleagues here on virological.org. After alignment with mafft (v4.790), we constructed a phylogenetic tree with iqtree (v1.6.12) with 1000 ultra-fast bootstrap replicates under the TIM+F+R3 substitution model (as estimated by ModelFinder). The tree was rooted to the earliest MPXV sequence (KJ642614) and visualized using R package ggtree (v3.15). The mafft alignment was visualized with snipit. The lollipop plot was generated from annotated vcf files with a slightly altered version of a previously developed R script.
Findings
For the swabs of patient 1 (Ct 22) and patient 2 (Ct 25) we generated 10 million and 8 million reads and achieved a coverage of 75x and 66x, respectively. For the virus isolate of patient 1, we generated 9 million reads of which 265k reads mapped to the reference genome achieving a mean coverage of 131x. We consistently detected 54 high-frequency variants compared to the MT903343 reference in all three samples.
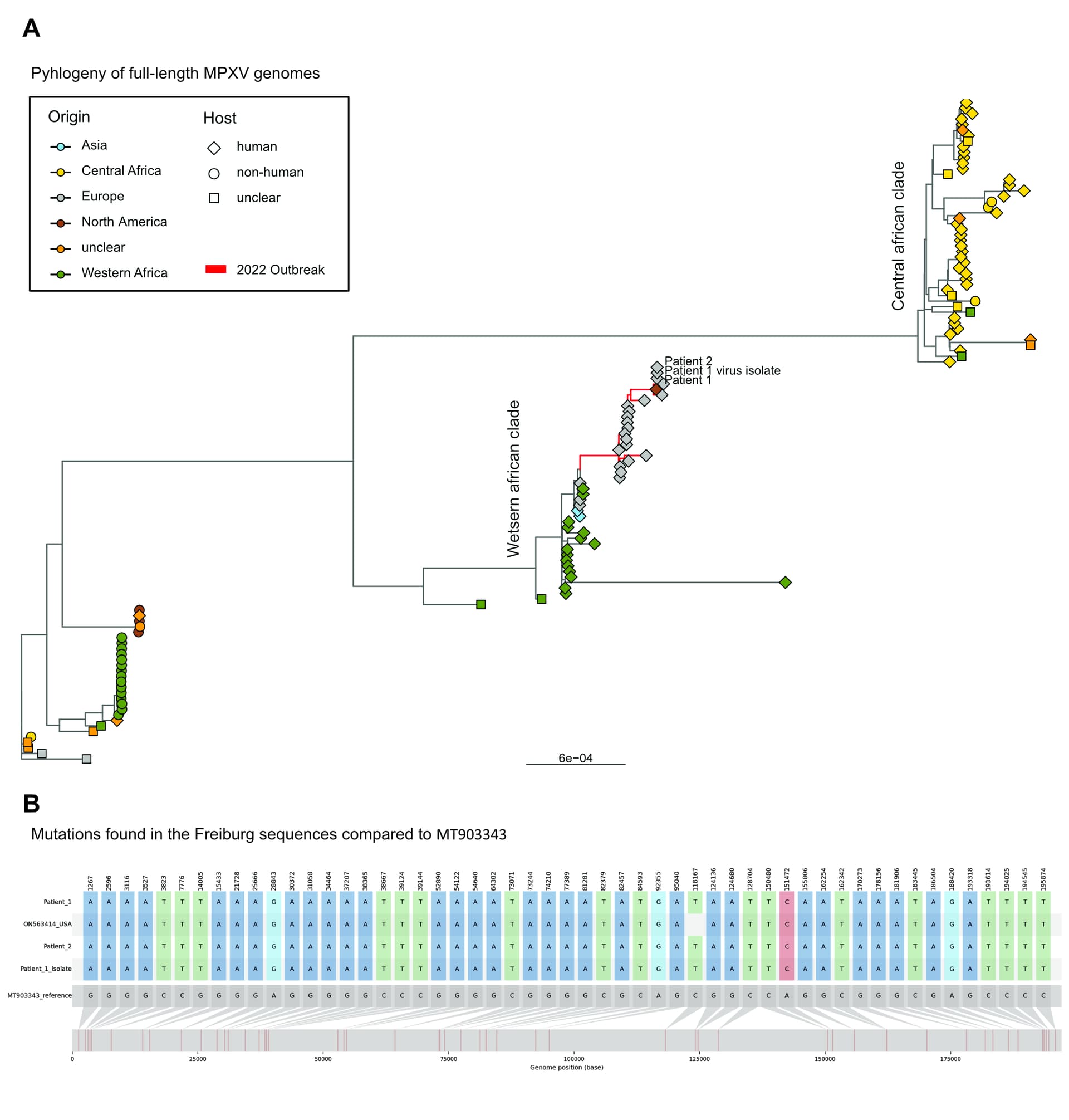
Phylogenetically, the samples clustered within the 2022 outbreak that is part of the western african clade (Figure 1A) and were most closely related to the 2022 US sequence ON563414 (Figure 1B). We also analysed the variant frequencies, including all variants significantly called over 5 % (Figure 2A) and show a high prevalence of non-synonymous mutations present in both patient sequences. Moreover, by analysing substitutions independent of the variant frequency, we observed that A and T variants occurred more often in both patients (Figure 2 B).

Although the significance of this finding has to be carefully evaluated, it might indicate an evolution of MPXV in humans possibly due to APOBEC3 deamination as already discussed here on virological.
The consensus sequences and raw data will be submitted to NCBI and ENA, but are available here for early access. We also included in this dataset the annotated vcf files and the output of iqtree.
References
[1] Di Giulio, D. B., & Eckburg, P. B. (2004). Human monkeypox: an emerging zoonosis. The Lancet infectious diseases, 4(1), 15-25.
[2] Adler, Hugh, et al. “Clinical features and management of human monkeypox: a retrospective observational study in the UK.” The Lancet Infectious Diseases (2022).
[3] Ladnyj, I. D., Ziegler, P., & Kima, E. (1972). A human infection caused by monkeypox virus in Basankusu Territory, Democratic Republic of the Congo. Bulletin of the World Health Organization, 46(5), 593.
[4] McCollum, A. M., & Damon, I. K. (2014). Human monkeypox. Clinical infectious diseases, 58(2), 260-267.