The ongoing evolution of variants of concern and interest of SARS-CoV-2 in Brazil revealed by convergent indels in the amino (N)-terminal domain of the Spike protein
Paola Cristina Resende 1a, Felipe G Naveca 2a, Roberto D. Lins 3, Filipe Zimmer Dezordi 4,5, Matheus V. F. Ferraz 3,6, Emerson G. Moreira 3,6, Danilo F. Coêlho 3,6, Fernando Couto Motta 1, Anna Carolina Dias Paixão 1, Luciana Appolinario 1, Renata Serrano Lopes 1, Ana Carolina da Fonseca Mendonça 1, Alice Sampaio Barreto da Rocha 1, Valdinete Nascimento 2, Victor Souza 2, George Silva 2, Fernanda Nascimento 2, Lidio Gonçalves Lima Neto 7, Irina Riediger 8, Maria do Carmo Debur 8, Anderson Brandao Leite 9, Tirza Mattos 10, Cristiano Fernandes da Costa 11, Felicidade Mota Pereira 12, Ricardo Khouri 13, André Felipe Leal Bernardes 14, Edson Delatorre 15b, Tiago Gräf 16b, Marilda Mendonça Siqueira 1b, Gonzalo Bello 17b, and Gabriel L Wallau 4,5b on behalf of Fiocruz COVID-19 Genomic Surveillance Network.
1. Laboratory of Respiratory Viruses and Measles (LVRS), Instituto Oswaldo Cruz, FIOCRUZ-Rio de Janeiro, Brazil.
2. Laboratório de Ecologia de Doenças Transmissíveis na Amazônia (EDTA), Instituto Leônidas e Maria Deane, FIOCRUZ-Amazonas, Brazil.
3. Department of Virology, Instituto Aggeu Magalhães, FIOCRUZ-Pernambuco, Brazil.
4. Departamento de Entomologia, Instituto Aggeu Magalhães, FIOCRUZ-Pernambuco, Brazil.
5. Núcleo de Bioinformática (NBI), Instituto Aggeu Magalhães FIOCRUZ-Pernambuco, Brazil.
6. Department of Fundamental Chemistry, Federal University of Pernambuco, Recife, Brazil
7. Laboratório Central de Saúde Pública do Estado do Maranhão (LACEN-MA), Brazil.
8. Laboratório Central de Saúde Pública do Estado do Paraná (LACEN-PR), Brazil.
9. Laboratório Central de Saúde Pública do Estado do Alagoas (LACEN-AL), Brazil.
10. Laboratório Central de Saúde Pública do Amazonas (LACEN-AM), Brazil.
11. Fundação de Vigilância em Saúde do Amazonas, Brazil.
12. Laboratório Central de Saúde Pública do Estado da Bahia (LACEN-BA), Brazil.
13. Laboratório de Enfermidades Infecciosas Transmitidas por Vetores, Instituto Gonçalo Moniz, FIOCRUZ-Bahia, Brazil.
14. Laboratório Central de Saúde Pública do Estado de Minas Gerais (LACEN-MG).
15. Departamento de Biologia. Centro de Ciências Exatas, Naturais e da Saúde, Universidade Federal do Espírito Santo, Alegre, Brazil.
16. Plataforma de Vigilância Molecular, Instituto Gonçalo Moniz, FIOCRUZ-Bahia, Brazil.
17. Laboratório de AIDS e Imunologia Molecular, Instituto Oswaldo Cruz, FIOCRUZ-Rio de Janeiro, Brazil.
a, b These authors contributed equally to this work.
Abstract
Mutations at both the receptor-binding domain (RBD) and the amino (N)-terminal domain (NTD) of the SARS-CoV-2 Spike (S) glycoprotein can alter its antigenicity and promote immune escape. We identified that SARS-CoV-2 lineages circulating in Brazil with mutations of concern in the RBD independently acquired convergent deletions and insertions in the NTD of the S protein, which altered the NTD antigenic-supersite and other predicted epitopes at this region. These findings support that the ongoing widespread transmission of SARS-CoV-2 in Brazil is generating new viral lineages that might be more resistant to neutralization than parental variants of concern.
Background
Recurrent deletions in the amino (N)-terminal domain (NTD) of the spike (S) glycoprotein of SARS-CoV-2 has been identified during long-term infection of immunocompromised patients 1–4 as well as during extended human-to-human transmission 3. Most of those deletions (90%) maintain the reading frame and cover four recurrent deletion regions (RDRs) within the NTD at positions 60-75 (RDR1), 139-146 (RDR2), 210-212 (RDR3), and 242-248 (RDR4) of the S protein 3. The RDRs that occupy defined antibody epitopes within the NTD and RDR variants might alter antigenicity 3. Interestingly, the RDRs overlap with four Indel Regions (IR) at the NTD (IR-2 to IR-5) that are prone to gain or lose short nucleotide sequences during sarbecoviruses evolution both in animals and humans 5,6.
Since late 2020, several more transmissible variants of concern (VOCs) and also variants of interest (VOI) with convergent mutations at the receptor-binding domain (RBD) of the S protein (particularly E484K and N501Y) arose independently in humans 7,8. Some VOCs also displayed NTD deletions like lineages B.1.1.7 (RDR2 𝚫144), B.1.351 (RDR4 𝚫242-244), and P.3 (RDR2 𝚫141-143) that were initially detected in the United Kingdom, South Africa, and the Philippines, respectively 3. The VOCs B.1.1.7 and B.1.351 are resistant to neutralization by several anti-NTD monoclonal antibodies (mAbs) and NTD deletions at RDR2 and RDR4 are crucial for such phenotype 9–14. Thus, NTD mutations and deletions represent an important mechanism of immune evasion and accelerate SARS-CoV-2 adaptive evolution in humans.
Several SARS-CoV-2 variants with mutations in the RBD have been described in Brazil, including the VOC P.1 15 and the VOIs P.2 16 and N.9 17. The VOC P.1 also displayed NTD mutations (L18F) that abrogate binding of some anti-NTD mAbs 14, but none of those variants displayed indels in the NTD. Importantly, although VOC P.1 showed reduced binding to RBD-directed antibodies, it is more susceptible to anti-NTD mAbs than other VOCs 9–14. In this study, we monitored and characterized the emergence of RDR variants within VOC and VOI circulating in Brazil that were genotyped by the Fiocruz COVID-19 Genomic Surveillance Network between November 2020 and February 2021.
Results
Our genomic survey identified 11 SARS-CoV-2 sequences from five different Brazilian states (Amazonas, Bahia, Maranhao, Parana, and Rondonia) that harbor a variable combination of mutations in the RBD (K417T, E484K, N501Y) and indels in the NTD of the S protein (Table 1). One VOI P.2 sequence and one VOC P.1 sequence displayed a convergent amino acid deletion 𝚫144 in the RDR2, while two VOC P.1 sequences displayed a four amino acid deletion 𝚫141-144 in the RDR2. On the other hand, one VOC P.1 sequence harbors a two amino acid deletion 𝚫189-190; two B.1.1.33(E484K) sequences carried deletions 𝚫141-144, 𝚫211 and 𝚫256-258, and four B.1.1.28 sequence displayed a four amino acid insertion ins214ANRN. We also identified B.1.1.28 ins214ANRN variants sharing six out of 10 P.1 lineage-defining mutations in the Spike protein (L18F, P26S, D138Y, K417T, E484K, N501Y) as well as P.1 lineage-defining mutations in the NSP3 (K977Q), NS3 (S253P) and N (P80R) proteins, thus defined as P.1-like variants. Inspection of sequences available at EpiCoV database in the GISAID (https://www.gisaid.org/) at March 1st, 2021, revealed one B.1.1.28 from the Amazonas state and three P.1 sequences from the Bahia state with deletion 𝚫144 (Table 1). All three P.1 𝚫144 sequences from Bahia were recovered from individuals reporting a travel history to the Amazonas state 18.
Table 1. SARS-CoV-2 Brazilian variants with indels at NTD of the Spike protein.
| Sample(s) | Lineage | NTD Indel | RBD | GISAID ID |
|---|---|---|---|---|
| AM-FIOCRUZ-20842572LS/2020* | B.1.1.28 | 𝚫144 | - | EPI_ISL_1068132 |
| MG-FIOCRUZ-8180/2021 | P.2 | 𝚫144 | E484K | EPI_ISL_1219137 |
| BA53/2021*, BA54/2021*, BA55/2021*, BA-FIOCRUZ-7029/2021* | P.1 | 𝚫144 | K417T E484K N501Y | EPI_ISL_1067729 EPI_ISL_1067733 EPI_ISL_1067734 EPI_ISL_1219136 |
| AL-FIOCRUZ-4795/2021, PR-FIOCRUZ-5273/2021 | P.1 | 𝚫144 | K417T E484K N501Y | EPI_ISL_1219134 EPI_ISL_1219133 |
| AL-FIOCRUZ-4786/2021* | P.1 | 𝚫189-190 | K417T E484K N501Y | EPI_ISL_1219135 |
| MA-FIOCRUZ-6871/2021, MA-FIOCRUZ-6874/2021 | B.1.1.33(E484K) | 𝚫141-144 𝚫211 𝚫256-258 | V445A E484K | EPI_ISL_1181371 EPI_ISL_1181370 |
| AM-FIOCRUZ-20897269OP*, AM-FIOCRUZ-20897281WS*, AM-FIOCRUZ-21840593CL*, PR-FIOCRUZ-5241/2021 | B.1.1.28 (P.1-like) | ins214ANRN | K417T E484K N501Y | EPI_ISL_1068256 EPI_ISL_1219132 EPI_ISL_1261122 EPI_ISL_1261123 |
| *Patient from *Amazonas state or traveler returning from Amazonas state. |
The Maximum Likelihood (ML) phylogenetic analyses showed that P.1 variants 𝚫141-144 were intermixed among non-deleted sequences (Fig. 1A). The four P.1 𝚫144 sequences detected in Bahia state; however, branched in a subclade (aLRT = 77%) together with the 𝚫189-190 variant and the other two lineages P.1 sequences that share the synonymous mutation A18945G (Fig. 1A). The four P.1-like ins214ANRN and the two B.1.1.33(E484K) 𝚫141-144/211/256-258 variants also clustered in highly supported (aLRT = 100%) monophyletic clades (Fig. 1A and B). These findings suggest that P.1 𝚫141-144 variants resulted from independent convergent NTD deletions events, while P.1 𝚫144, P.1-like ins214ANRN and B.1.1.33(E484K) 𝚫141-144/211/256-258 variants might represent newly emergent VOIs or VOCs. It is interesting to note that most P.1 sequences with NTD deletions were detected in individuals from or with travel history to the Amazonas state.
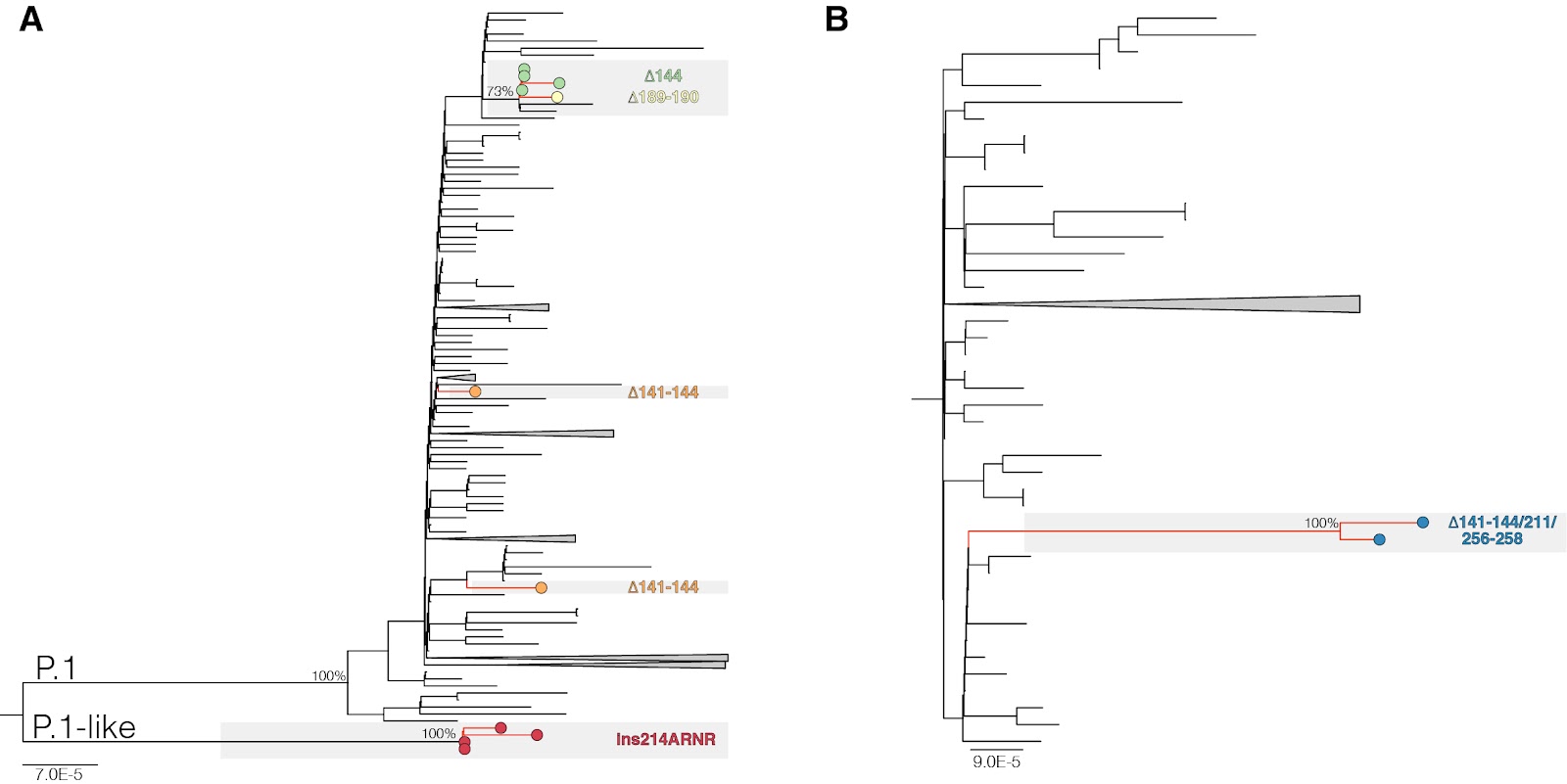
Figure 1. ML phylogenetic tree of whole-genome lineage P.1/P.1-like (A) and B.1.1.33 (B) Brazilian sequences showing the recurrent emergence of deletions at the NTD of the S protein. Tip circles representing the SARS-CoV-2 sequences with NTD indels are colored as indicated. The trees were rooted at the midpoint and branch lengths are drawn to scale with the left bar indicating nucleotide substitutions per site. For visual clarity, some clades are collapsed into triangles.
While SARS-CoV-2 variants harboring NTD deletions at RDR2 and RDR4 have emerged in many different lineages globally, the ins214 in the S protein is a more rare event. Our search of SARS-CoV-2 sequences available at EpiCoV database in the GISAID (https://www.gisaid.org/) at March 1st retrieved only 146 SARS-CoV-2 sequences of lineages A.2.4 (n = 52), B (n = 3), B.1 (n = 7), B.1.1.7 (n = 1), B.1.177 (n = 1), B.1.2 (n = 1), B.1.214 (n = 80) and B.1.429 (n = 1) that displayed an insert motif of three to four amino acids (AKKN, KLGB, AQER, AAG, KFH, KRI, and TDR) in position 214 (Appendix Table 1.pdf (13.2 KB). Most ins214 motifs were unique, except ins214TDR, which seems to have arisen independently in B.1 and B.1.214. With the only exception of one lineage B sequence sampled in March 2020, all SARS-CoV-2 ins214 variants were only detected since November 2020, and its frequency increased in 2021 mainly due to the recent dissemination of lineage A.2.4 ins214AAG in Central and North America and lineage B.1.214 ins214TDR in Europe.
Next, we aligned the S protein of representative sequences of SARS-CoV-2 lineages with NTD indels and SARS-CoV-2-related coronavirus (SC2r-CoV) lineages from bats and pangolins 19. Inspection of the alignment confirms that most NTD indels detected in the SARS-CoV-2 lineages occur within IR previously defined in sarbecovirus (Fig. 2). The 𝚫141-144 occurs in the IR-3 located in the central part of the NTD, where some bats SC2r-CoV also have deletions. The ins214 occurs in the IR-4 where an insertion of four amino acids was detected in three bat SC2r-CoV isolated in China (RmYN02, ins214GATP), Thailand (RacCS203, ins214GATP), and Japan (Rc-o319, ins214GATS). Despite amino acid motifs at ins214 were very different across SARS-CoV-2 and SC2r-CoV lineages, the insertion size was conserved (3-4 amino acids). The 𝚫256-258 occurs near the IR-5, where some bat and pangolin SC2r-CoV lineages also displayed deletions. Thus, the NTD regions that are prone to gain indels during viral transmission among animals are the same as those detected during transmissions in humans.
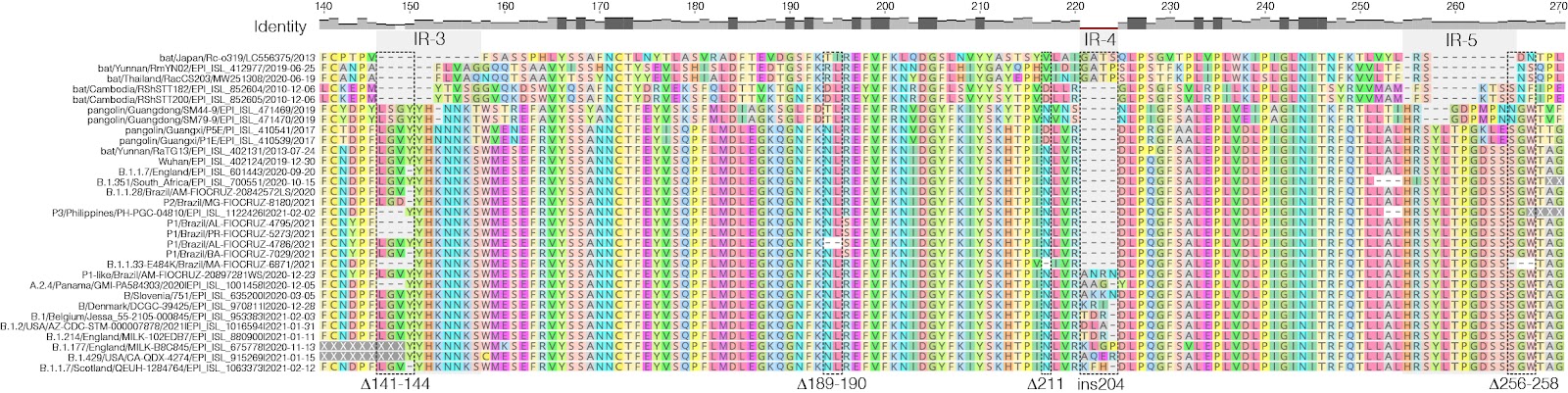
Figure 2. Amino acid alignment of positions 140-270 of the S protein of representative sequences of SARS-CoV-2 lineages harboring indels in the NTD and SARS-CoV-2-related coronavirus (SC2r-CoV) from bats and pangolins. IRs positions (gray shaded areas) are approximations due to the high genetic variability in these alignment positions. Dotted rectangles highlight the indels identified in this study. The identity level estimated for each position of the alignment is displayed at the top.
Epitope mapping showed that neutralizing antibodies are primarily directed against the RBD and NTD of the S protein 9,20–23. Some of the RBD mutations (K417T and E484K) detected in the VOCs and VOIs circulating in Brazil have been associated with increased resistance to neutralization by mAbs, or polyclonal sera from convalescent and vaccinated subjects 24–27. The RDR2 𝚫144 and RDR4 𝚫242-244 deletions observed in VOCs B.1.1.7 and B.1.35, respectively, are located in the N3 (residues 141 to 156) and N5 (residues 246 to 260) loops that composes the NTD antigenic-supersite 28,29 and confers resistance to neutralization by anti-NTD mAbs 3,9,10,30. Moreover, in vitro co-incubation of SARS-CoV-2 with highly neutralizing plasma from COVID-19 convalescent patient, has revealed an incremental resistance to neutralization followed by the stepwise acquisition of indels at N3/N5 loops 31. Notably, SARS-CoV-2 challenge in hamsters previously treated with anti-NTD mAbs revealed a selection of two escape mutants harboring NTD deletions 𝚫143-144 and 𝚫141-144 14. Thus, NTD indels might represent a mechanism of ongoing adaptive evolution of VOC and VOI circulating in Brazil to escape from dominant neutralizing antibodies directed against the NTD antigenic-supersite.
To test this hypothesis, we performed a modeling analysis of the binding interface between wildtype/indels NTD variants and the NTD-directed neutralizing antibody (NAb) 2-51 derived from a convalescent donor 20. The NAb 2-51 makes several contacts with the wildtype NTD antigenic-supersite (EPI_ISL_402124), primarily through the heavy-chain (Fig. 3). The loops N3 and N5 play a pivotal role in the binding process with a predominance of hydrophobic contacts and dispersion interactions in N5 and saline interactions in N3. Our result shows that deletions at RDR 2 (𝚫144, 𝚫141-143) and RDR4 (𝚫242-244) impact the loops’ size and conformation, disrupting the native contacts and reducing the interacting hydrophobic surface accessible area, mainly due to the loss of the hydrophobic pocket (Figure S1.pdf (276.8 KB)). Indels around the N3/N5 loops resulted in a significant loss of interactions (both electrostatic and hydrophobic) (Table 2) that will dramatically impact the binding free energy, and therefore the binding affinity, between those NTD deletion variants and the NAb 2-51. Although NTD indels 𝚫189-190 and ins214ANRN did not affect the NTD antigenic-supersite, they occur at putative epitope regions covering residues 168/173-188 and 209-216 (Appendix Table 2.pdf (62.8 KB) ) and leads to conformational changes in exterior loops (Figure S1 G-H) which might affect Ab binding outside the antigenic-supersite. These findings suggest that NTD deletions 𝚫144, 𝚫141-143, and 𝚫242-244 here detected probably abrogate the binding of NAb directed against the NTD antigenic-supersite and confirm that deletions at RDRs 2/4 are an essential mechanism for SARS-CoV-2 immune evasion 3,14.

Figure 3. List of native interactions showed onto the 3D structure of the S protein NTD targeted by a natural mAb. Cartoon representation of the structure of the NTD protein complexed to the NAb 2-51. The NTD is colored in pink; the heavy and light chains of the NAb 2-51 are colored in gray and green, respectively. The insets show a close-up of the binding interface of the loops N3 and N5 interacting with the variable chains of the NAb 2-51. The N5 loop representation is also rotated 180° around its z-axis. Residues making contact in the interface are depicted in the licorice representation, with carbon atoms in cyan, nitrogen atoms in blue and oxygen atoms in red. The dotted lines indicate the interacting residues-pair.
Table 2. Impact of indels on the binding between SARS-CoV-2 NTDs and NAb 2-51, expressed as loss of putative interactions.
| Variant | ΔH-bond | ΔSalt-bridge | Δpi-stacking | ΔHydrophobic SASA [Å2] | Native Contacts Lost (NTD - Ab) |
|---|---|---|---|---|---|
| P.1 𝚫189-190 | 0 | 0 | 0 | 0 | - |
| P.1-like ins214ANRN | 0 | 0 | 0 | 0 | - |
| B.1.1.28 𝚫144 | -2 | -3 | -1 | -1 | K147-E71, K150-E53, K150-D54, Y145-Y98 |
| P.2 𝚫144 | -2 | -3 | -1 | -104 | K147-E71, K150-E53, K150-D54, Y145-Y98 |
| P.1 𝚫144 | -2 | -3 | -1 | -111 | K147-E71, K150-E53, K150-D54, Y145-Y98 |
| P.1 𝚫141-144 | -2 | -3 | -1 | -313 | K147-E71, K150-E53, K150-D54, Y145-Y98 |
| B.1.1.33 𝚫141-144 𝚫256-258 | -3 | -3 | -1 | -439 | Y147-E71, K150-E53, K150-D54, Y145-Y98, D253-S56, P251-P55, P251-L46, P251-Y100 |
Recent genomic findings are showing a sudden landscape change in SARS-CoV-2 evolution since October 2020, coinciding with the independent emergence of VOCs carrying multiple convergent amino acid replacements at the RBD of the S protein 32. One hypothesis is that such a major selection pressure shift on the virus genome is driven by the increasing human population immunity worldwide acquired from natural SARS-CoV-2 infection. Our findings suggest that SARS-CoV-2 is continuously adapting in Brazil and that RDRs 2/4 variants here detected might have evolved to escape from NAb against NTD supersite and could be even more resistant to neutralization than the parental P.1, P.2, and B.1.1.33(E484K) viruses. The sequential evolution steps observed in Brazil recapitulates the pattern observed in South Africa where the VOC B.1.351 first acquired key RBD mutations (E484K and N501Y) and some weeks later the NTD deletion 𝚫242-244 7. These findings highlight the urgent need to address the SARS-CoV-2 vaccines’ efficacy towards those emergent SARS-CoV-2 variants and the risk of ongoing uncontrolled community transmission of SARS-CoV-2 in Brazil for the generation of more transmissible variants. Furthermore, the recurrent emergence of NTD ins214 variants in different SARS-CoV-2 lineages circulating in the Americas and Europe since November 2020 deserves further attention.
References
41 Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph 1996; 14: 33–38.