I have been privately dealing with rumors and inquiries, focused on the RRAR potential furin cleavage site, that nCoV2019 may have a suspicious origin as an engineered, laboratory-generated virus either accidentally or deliberately released in the area of the Wuhan seafood and animal market. The publication of the highly similar RaTG13 sequence about a week ago has fueled this type of speculation.
As I have told people privately, I see no evidence at all to support such a claim. In sharp contrast, I have studied the question in detail, using RaTG13 and Wuhan sequence at the S1/S2 boundary, and find convincing proof of exactly opposite conclusion – that RaTG13 could NOT be a proximal source of the Wuhan virus.
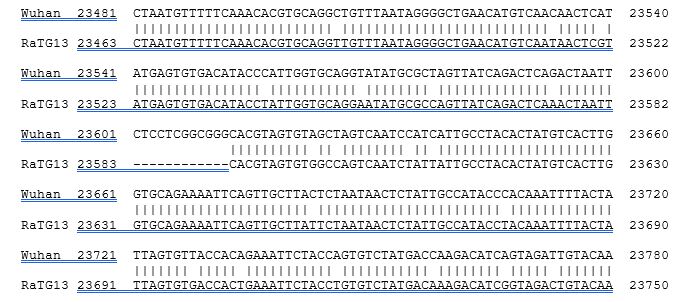
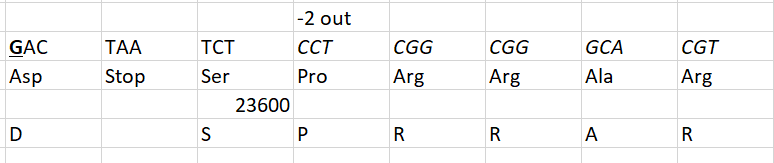
At first glance of an alignment of the S protein sequence of both, it is natural that the issue of an engineered insertion should be considered. On either side of the new furin site, the amino acid sequence is identical in both from aa614 to aa1133 – an apparent insert of PRRA is the only difference in an otherwise 100% conserved 519 amino acid region.
One has to consider that the PRRA is an unusual sequence to introduce to generate a furin site – others even among coronaviruses like MHV A59 are so much better. Also that the underlying code CCTCGGCGGGCA introduces an unnecessarily G and C rich region where none otherwise exists. Not likely scenarios for something a gene jockey would do.
Then one looks at the actual RNA alignment. The “insert” is actually not in frame, but CTCCTCGGCGGG, or -2 out of frame. Again, who does that?
But the PROOF lies in looking at the 288 alignable nucleotides on either side of the “insert”. While they cover identical protein sequence, the RNA is not at all identical, but 6.6% different – 19 mutations out of 288. All 19 are mutations in the wobble base of their respective codons. There are so many that the frame can be inferred from the 2/1 pattern even without knowing the beginning or the end, or indeed that the encoded protein sequence is identical – those are self-evident by looking at the RNA itself.
We know from influenza H1N1, for which we have serial isolates from 1918 to the present, that wobble base mutagenesis occurs at a rate of 0.95% per decade. This permits an estimation of the TMRCA of the two sequences nCoV2019 and RaTG13 of 69.5 years ago – roughly 1950 +/- 10 years or so.
RaTG13, or anything nearly identical to it at the RNA level, simply could not be a proximal source of nCoV2019. It just LOOKS like it might be…at first glance.
Given that furin cleavage signals are present in other coronaviruses at exactly that point in the S1/S2 boundary region, it only LOOKS unusual, especially against the backdrop of SARS. The preponderance of evidence, coupled with Ockham’s razor (that the simplest explanation is preferred) dictates that the PRRA sequence has been conserved in nCoV2019 from a long ago ancestor virus. It is not of suspicious origin. The closest bat virus sequence is really not close at all.
You have it framed incorrectly, omitting a T, to wit, from Genbank entry:
23581 ttatcagact cag act aat tct cct cgg cgg gca cgt agt gtagctagtc aatccatcat
So no stop.
Also, it is the third base, the wobble base, that is consistently mutated over time in a highly conserved, constant amino acid sequence.
I did make an arithmetic error, pointed out to me by email – the TMRCA is 38.5 years between Bat RaTG13 and the novel coronavirus. Still far too long a time for Bat RaTG13 to be a proximal source.
I stand by my analysis.
SOURCE OF FURIN SITE IN SARS-CoV-2 IS COPY CHOICE ERROR FROM MIXED INFECTION OF BATS
I have found a probable source of the putative insert that adds the RRAR furin site to SARS-CoV-2.
Ten of the 12 nucleotides in the RRAR insert are identical to a sequence in the spike protein gene of Bat Coronavirus HKU9 isolated from a Rousettus fruit bat in Guangdong province in 2011.
In HKU9 they code for TSAG in the loop region known in the Gao terminology as SD3. The first CT of the putative insert is still orphan, but there is a relevant possible source for the rest, albeit out of frame.
The “Insert” is CT CCTCGGCGGG, the last ten identical to the sequence in HKU9.
The sequence context has other similarities, i.e the “cagac” upstream and a “c” downstream of the “insert” as aligned here:
HKU9 gcatttgta caga------cctcggcgggc ctctgt
CoV-2 tatcagact cagac ttgct cctcggcgggc acgtagt
Total of 14 of 19 identical nucleotides in two runs with one gap. That’s a lock.
Copy choice errors in mixed infections do not have to be in an exact context, since it is not a recombination, but RNA polymerase hopping from one template strand to another. In this case the two templates code for the same gene, between regions about a thousand nucleotides from one another, out of a genome of 30,000 nucleotides. There could easily have been evolution of the site since the mixed infection, over a period of years.
This cannot be a coincidence.
The natural range of the host for Bat RaTG13, closely related to SARS-CoV-2, the microbat Rhinolopus affinis, overlaps that of the Rousettus fruit Macrobat in Yunnan province. The latter is used for bush meat. Also, Microbats and Macrobats are known to co-habit the same caves. An alternate source of virus for a human infection could be mixed bat guano from such a cave with a mixed population.
So the definitive source of the pandemic is a mixed infection of viruses similar to SARS-CoV-2 and Bat HKU9 – copy choice error resulting in an insert in SARS-CoV-2. Could occur in bats, intermediate animal or human.
Human in incubation with SARS-CoV-2 could hop a max speed train, direct line from Kunming in Yunnan Province and disembark in Wuhan train station 6.5-8 hours later, sometime last fall. The line began service in 2017.
Infected in Yunnan, does not spread it there, but goes to Wuhan, where he/she either falls ill or spreads it asymptomatically to another person, initiating the pandemic there. The initial outbreak occurs a short distance from the Wuhan train station for good reason – that is where it arrived.
This accidental mixed infection in the wild, and infection of a human by some form of bush meat or bat guano, who carries it by high speed train to Wuhan – this is the most likely series of events leading to the pandemic.
The only laboratory required is the natural laboratory of the bat cave with multiple species of bats and bat coronaviruses.
Found it.
William R. Gallaher. Ph.D.
Professor of Microbiology, Immunology and Parasitology, Emeritus
The fact that that area of the SARS-CoV-2 genome is a site of RNA polymerase stuttering is clear from the tandem repeat in the RNA sequence, that evidently occurred in the long ago ancestor of SARS-CoV-2 and Bat RaTG13 (tandem repeat mutated in latter) just upstream of the site of the insert, thusly,
SARS-CoV-2 tat cagact cagact tgctcctcggcgggcacgtagt
This is, after all, a looped out peptide sequence, and even when the furin site is common, as in other Coronaviruses, there is not strict alignment relative to neighboring, more constant, peptide regions. The irony of a functionally important loop that is also hypervariable.
This is a really nice finding, Bill. This sequence is in lots of things when you BLAST it but the fact it is in a bat CoV is very convincing to me that that is the source. Adds to the evidence that the polybasic insertion occurred in a bat and not some other animal.
I’m new to this forensic analysis, and not expert on protein design, so please forgive any naivety. But if you were doing something like this for evil purposes, wouldn’t you throw down a bunch of synonymous changes too, to make it look like something older (and thus less traceable)?
I didn’t really mean anything to do with labs. There is still a question as to whether this insertion arose in an intermediate host (or potentially in cryptic transmission in humans). This observation increases the likelihood that the insertion occurred in a bat and thus SARS-CoV-2 was a fully human transmissible virus in the bat species.
The sequence analysis all seems very cogent, especially the discussion on copy-choice errors. But I’m a bit confused about the geographical aspect of your supposition. You say that the HKU9 CoV was found in the Guangdong province, but your transport to Wuhan involves a train from Yunnan. Does that mean you suppose that the mixed infection was in the intersection of Rousettus and Rhinolopus ranges, and then the novel sequence was spread unidirectionally to Yunnan (e.g., Kunming)? Is it not possible that it was maintained in the macrobat, spreading to points in Guangdong that are accessible to/from Wuhan? That seems a little closer. As for the carrier being a human, it would seem surprising to me that this person could be the only one who handled the animal in question and that s/he became contagious only shortly after disembarking from the hypothetical train. I wonder if bats and other animals are themselves transported long distances in China to fill the demand in various markets. Are there bushmeat brokers who might engage in that sort of shipment?
You reference Occam’s Razor in some of your writing. I’m a fan too, so I’m trying to overcome the skepticism that bubbles up in my mind regarding this sole-traveler hypothesis…
I suppose one of the things that was always attractive about the pangolin hypothesis is that this animal is rare enough to be transported long distances. My intuition says that is probably not the case for bats.
We have too ample evidence now that the animal that can transport SARS-CoV-2 from point A to point B, and spread it the fastest is a human being. Caged animals of any kind need not apply – and no animal reservoir for the virus has been found, now several months into the investigation. It is not for lack of the Chinese looking - they are far more motivated to understand the genesis of this than anyone.
The "pangolin hypothesis’ was based on faulty molecular analysis of sequence similarity, which turned out to not be that similar at all. That explanation was debunked 2.5 months ago.
I am not a fan of “gain of function” experiments at all, but the work of Zhengli and colleagues has shown that direct bat to human Coronavirus infection is possible. We are certain of it for Ebola.
The overall sequence of SARS-CoV-2 is closest to Yunnan isolates – only a bit appears to come from a virus that is HKU9-like. So it most likely came from Yunnan – but the outbreak occurred 700 miles away in Wuhan, not in Yunnan. Not along the way there either. The sole human traveler on high speed train while still in incubation is the best fit to the known information, in my view. Ockham’s razor (note spelling).
Microbats, Macrobats and Bat Coronaviruses have been circulating through the Karst limestone formations in the south of China, that stretches from southeast Yunnan to western Guangdong, far longer than the Chinese people have been in existence. That is saying a lot, but it is a truth attested to by the very wide varieties found there of both hosts and viruses…Human Coronavirus disease out of that environment has been an accident waiting to happen – and still is. Emergence happens.
About an hour from this writing, I can go on my porch and watch a flight of microbats rise from hollow trees in the wetland forest at the rear of my property, to skim the headwaters of Gum Creek for their nightly meal of insects. It is the peril of being a virologist that there are times I wonder about their virome.
But right now, the grocery store is a far more dangerous place than my forest. We would all instinctively like to blame that on someone, somewhere. The truth is that every virus of human beings represents a past accidental emergence, an innocent wrong place, wrong time, thing.
The entirely natural origin of SARS-CoV-2 is as simple as CAGAC.
That palindrome, or its complement TGAGT, or the variant CAGAT, very frequently precedes a length polymorphism or copy-choice recombination event among Bat Coronaviruses.
Palindromes act like speed bumps for the RNA polymerase complex, rather like switches in a train yard. The polymerase can stutter or jump to another track, perhaps to jump back, or not.
Swapping out RNA sequences in bat caves is as simple as CAGAC. That in a nutshell is how this all happened.
Regarding the overall similarity to Yunnan sequences: OK, fair enough. But frankly, this just further tips my intuition toward an accidental escape from the Wuhan lab. It seems there is a lot of research activity devoted to collecting viruses in Yunnan. And, in keeping with your personal account of the critters inhabiting your local environs (which I do not think of as replete with Karst geology; correct me if I’m wrong), I keep finding myself wondering… Why do the researchers’ collection sites emphasize locations so far from Wuhan? Surely there must be bats in and around Wuhan. Is it simply the case that most of the coronaviruses that can infect both bats and humans come from the Yunnan region?
Regarding the spelling of Fra. William of Ockham: I wonder why references to the “razor” so frequently use the “Occam” spelling? I suppose it has to do with the Latin phrase “novacula occami” recorded by Libert Froidmont. And there we arrive at a dilemma: If William merely expressed a philosophy without explicitly invoking the steely metaphor, perhaps the naming rights should go to Prof. Foidmont. (In fact, it seems there is little in William’s surviving writings that really justify singling him out for edgy parsimony, related antecedents going back at least as far as Aristotle.) Nevertheless, if English-speakers are going to translate “novacula” to “razor,” it would only seem consistent to restore what is considered to be the English spelling of William’s birthplace. Thanks for prompting me to look into the matter.
A COMMON PALINDROMIC RNA SEQUENCE AS UNITARY CONTRIBUTOR TO COPY-CHOICE RECOMBINATION IN SARS-COV-2
This is intended to be in the same vein as my original post in this thread, just before midnight of Feb 6, evaluating similarities in Coronavirus sequences at the level of viral RNA. In this case, the subject is the observation that the receptor-binding domain of SARS-CoV-2 bears significantly high similarity to that of a Coronavirus recently obtained from a pangolin, namely Pan_SL-CoV_GD/P1L.
I have hesitated to comment, because an extensive analysis of this issue has been posted as a pre-review manuscript on the preprint website bioRxiv since March 24, and my preference would have been to wait for its publication, along with the posting of the Pan_SL-CoV_GD/P1La sequence on Genbank. It is now 7 weeks later, and neither the paper nor sequence have been posted on PubMed or Genbank. In this era of rapid publication, especially for COVID-related work, this is highly unusual.
The current citation is:
Emergence of SARS-CoV-2 through Recombination and Strong Purifying Selection
Xiaojun Li, Elena E. Giorgi, Manukumar Honnayakanahalli Marichann, Brian Foley, Chuan Xiao, Xiang-Peng Kong, Yue Chen, Bette Korber, Feng Gao
bioRxiv 2020.03.20.000885; doi: https://doi.org/10.1101/2020.03.20.000885
I will begin by saying I concur completely with this paper, by a team of authors I hold in high regard. I have also communicated directly with Brian Foley of the team several days ago. I wish only to add additional information consistent with my previous posts on this thread, and not “republish” their work in any way.
Likewise, colleagues of mine in a global collaboration posted here that SARS-CoV-2 was not derived from any pangolin sequence, in:
I concur completely with their analysis, subsequently published in Nature Medicine, done largely at the amino acid level.
I wish here to compare Bat RaTG13 and Pan_SL-CoV_GD/P1L with SARS-CoV-2 principally at the level of viral RNA sequence, to show how a common palindromic RNA sequence may be the unitary contributor to several events of copy-choice recombination that gave rise to these viral sequences.
The Pan_SL-CoV_GD/P1L sequence I used is the original incomplete RNA sequence, first described in:
Lam, T.T., Shum, M.H., Zhu, H. et al. Identifying SARS-CoV-2 related coronaviruses in Malayan pangolins. Nature (2020). Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins | Nature
While incomplete, the gaps do not affect the areas of sequence to be discussed here. I will use the DNA equivalent, derived by reverse transcription, as is common practice.
The central point of the relationship between the receptor-binding domain (RBD) among the three viral sequences can be seen in this amino acid alignment, derived from the much more complex Figure 2A of the Li et al. 2020 paper, to wit:
Amino acid changes are highlighted in blue This makes their point that the RBD of SARS-CoV-2 is not derived from a virus similar to Bat RaTG13, but rather from one similar to a virus derived from pangolin.
The authors do briefly allude to a much greater difference in RNA sequence between SARS-CoV-2 and the pangolin virus, but I would submit that a closer look at the nature of that difference should be made more clearly. What follows is an annotated alignment of the RNA sequences in this region, from each of the three viruses.
As I first posted concerning the relatedness at the RNA level between Bat RaTG13 and SARS-CoV-2, this alignment is replete with wobble base mutations (blue arrows), between SARS-CoV-2 and Pan_SL-CoV_GD/P1L. There are 28 in all over a span of 268 nt between apparent changes of track from RaTG13-like to Pan_SL-CoV_GD/P1L-like and back to RaTG13-like sequence.
We know from many other virological examples that it takes several decades to accumulate this level of wobble-base mutagenesis, as I described Feb 6 in my first post to this thread. In this case, my estimate would be divergence over a span of 40 years.
So the recombination event resulting in this RBD sequence being in SARS-CoV-2 occurred in a decade around 1980. Not only could this not have occurred in a lab, but it is also unlikely to have occurred in a pangolin.
Pangolins are solitary animals, meeting only to mate. They are very unlikely to be capable of horizontal transmission of a virus, about as unlikely as hermits living in the wilderness. Rather, they reflect transmission from bats within their range of habitation. So the copy-choice recombination event that led to SARS-CoV-2 having an RBD sequence capable to binding to the ACE-2 receptor occurred in a bat cave four decades ago.
It is also worthy of note that the RNA pentanucleotide CAGAT, a variant of CAGAC that I highlighted in a recent post, lies directly before the likely area of crossover in the recombinant.
So, the two most unique peptide sequences of SARS-CoV-2 related to its ability to infect human beings and spread rapaciously, the RBD and furin cleavage site, are unified by being preceded by the CAGAC/CAGAT motif.
There are other nearly identical sequences, exceeding 99% at the RNA level, noted by the Li et al and Lam et al papers: the coding sequence for Membrane Protein E and for the 3’OH terminus downstream of the nucleocapsid (N) gene. In all three viral sequences for Membrane Protein E, as well as SARS of 2003 (that is identical in the first 50 amino acids with these other three), the palindrome TGAGT is found, which is the complement to CAGAC,just prior to the E gene. Finally, as shown below, the beginning of the 3’OH RNA sequence, identical in all three viruses, is replete with five nucleotide palindromes, including CAGAC and its variant CAGAT.
Even in SARS of 2003, the sequence identity in the 3’OH region is 3%, far lower than the overall 20% disparity between SARS and SARS-CoV-2.
Therefore, as shown below, much of the critical evolutionary history of both SARS and SARS-CoV-2 can be associated with the proximity of copy-choice recombination sites to CAGAC, its complement, or a similar pentanucleotide.
Others have noted the profligacy of recombination sites within the coronavirus genome that have accumulated over their very long evolutionary history in bats. So there may well be other RNA sequence motifs that tend to facilitate copy-choice errors.
With respect to SARS-CoV-2 and the ancestral viruses that contributed critical regions to its RNA sequence for human pandemic potential, this was clearly a natural process. This reflects an evolution over decades, in bat caves long ago, facilitated by some mechanism, as shown above, whereby CAGAC disrupts the processivity of the viral RNA polymerase complex down its template, and facilitates, albeit rarely, copy-choice errors capable of creating potentially dangerous recombinants to humankind.
To date, no source of SARS-CoV-2 has been determined, and neither bat, nor other mammal, has been found to harbor it except human beings in the pandemic.
All around the globe, those of us who have studied emerging viral pathogens at the molecular level for decades are united in our judgment, based on protein and RNA sequence analysis, that SARS-CoV-2 evolved by a series of recombination events in the wild. Sequence divergence shows that these events occurred through many decades of recombination among both similar and distantly-related bat Coronaviruses, potentially in multiple bat species co-habiting in the same limestone bat caves across a wide swath of southern China.
The reader will note that the sequences are labeled “pangolin”, in parentheses, Pre-SARS, Pre-SARS-CoV-2, Pre-Bat RATG13, Pre- Pan_SL-CoV_GD/P1L, and Pre-HKU9. This is because the source viruses come from the past, and not the present. From past locations, and not the location or host species from which they happened to be much more recently isolated.
This judgment is based on facts and molecular evidence, independently judged by different analyses in the hands of eminently qualified scientists. A number of us have never worked together over long careers. Some of us do not even know of each other by reputation. Yet we have all come to the same conclusion, in China, in Scotland, in North Carolina, in Louisiana, in Texas, in New Mexico, in California and in Australia. The backbone of the virus sequence was derived from a common ancestor of Bat RaTG13 and SARS-CoV-2, most likely in Yunnan province from which Bat RaTG13 was isolated. Small segments of sequence, hundreds of nucleotides long in a genome of 30,000 nt, were derived from viruses ancestral to other viruses only recently isolated in Guangdong province.
The only laboratory in which SARS-CoV-2 was concocted was a natural one in a bat cave, in a process that took decades, an accident of nature waiting for human contact.
William R. Gallaher, Ph.D. (Harvard ’72)
Professor of Microbiology, Immunology and Parasitology, Emeritus
Louisiana State University School of Medicine, New Orleans
I read with much interest this thoughtful thread; thanks to you all. I’ve been studying the evolution of the spike protein in detail as well.
Two issues are still open (not only) in my view: 1) the origin of nCoV-19 and 2) the origin of the furin cleavage site.
The origin of nCoV-19.
The physicist "John von Neumann used to say, with four parameters I can fit an elephant, and with five I can make him wiggle his trunk.” (quoted by Enrico Fermi, in Dayson F. Nature 2004; 427:297). Now, given that analyse Chinese railways’ timetables is far beyond my skills, I’d go instead for something more traditional: get more data. Trivial? Maybe not. I am deeply astonished by how much has been said and published on the origin(s) of nCoV-19, and all of that mainly based on the comparisons with a single sequence: Bat-CoV-RaTG13. How could this have happened?
I am used to teach students that for meaningful phylogenetic/evolutionary analyses, taxon sampling should be as balanced a possible. Here, bat relatives of the Bat-CoV-RaTG13 are clearly underrepresented. Proposed solution: Someone should ask Dr Zheng-Li Shi, where she collected that precious sample. Then send someone over there, collect some more bat samples and end once and for all this story without the need for more just-so stories. Anybody agrees with my proposal?
The origin of the furin cleavage-site.
In this thread the main proposal about the origin of the furin cleavage-site invokes replication errors and the CAGAC palindrome. Is there a chance that Gallaher’s “RNA polymerase stuttering” hypothesis could be tested in the lab? That would be terrific.
Yet, one thing is to generate (synonymous) mutations, a quite different one is to get them fixed in the population.
Yes, the S1/S2 cleavage site is present in other Coronaviruses (eg. ref to Table1-Fig2, in Coutard et al., 2020, Antiviral Res, https://doi.org/10.1016/j.antiviral.2020.104742). But not quite the same site as in 2019-nCoV, as pointed out in Walls et al. (Cell 2020, 180, 1–12). This cleavage site is “conserved among the 144 SARS CoV-2 isolates sequenced to date but not in the closely related RaTG13” (Walls 2020). The Authors demonstrate that the acquisition of the RRAR recognition site in 2019-nCoV is" something special". Indeed, the Western blot in Fig 1D shows that the S1/S2 cleavage takes place in 2019-nCoV but not in SARS-CoV: the two cleavage site are therefore not “equivalent”. Nor the cleavage takes place in a mutated sequence, in which only the Arg685 is conserved “… thereby mimicking the S1/S2 cleavage site of the related SARSr-CoV S CZX21” (Walls 2020).
Establishing the origin of the S1/S2 cleavage site in 2019-nCov is imperative for “… it is becoming increasingly apparent that proteolytic activation of spike by host cell proteases also plays a critical role … in cell and tissue tropism, host range, and pathogenesis” (Millet & Whittaker, 2015, Virus Res 202:120-134) … and “… S-protein “priming” … may provide a gain-of-function to the 2019-nCoV for efficient spreading in the human population compared to other lineage b beta-coronaviruses” (Coutard et al., 2020, Antiviral Res, https://doi.org/10.1016/j.antiviral.2020.104742).
Conclusion: To me, the origin of the furin cleavage-site in 2019-nCov is not clear yet.
Multiple peer-reviewed articles have shown that cats are susceptible to SARS-CoV-2, which raises an interesting question - could a SARS-CoV-2 ancestor without PRRA infect cats and recombine with FIPV to acquire PRRA? Could this be tested experimentally?
Hi Fuyutao, can you share the GenBank or GISAID accession no. of those feline coronavirus with the S1/S2 PRRA? I tried searching and there are 1000s of hits.
I have submitted a paper entitled “A COMMON PALINDROMIC RNA SEQUENCE AS UNITARY BREAKPOINT CONTRIBUTOR TO COPY-CHOICE RECOMBINATION IN SARS-COV-2” and posted a preprint version (subject to revision post-review) on bioRxiv. I will post the doi when available.
Relevant to the breakpoint sequence hypothesis are the number and position of CAGAC and CAGAT on the plus and minus strands of SARS-CoV-2. A map of these is not included in the paper, due to its file size, but a pdf copy is made available here.
The map not only indicates the location of breakpoint sequences, but also in relation to the RNA code for the known gene products of the virus. Navigation through the 30K genome as challenging, and I hope this helps.
I have run a similar breakpoint analysis on the ref seq for Middle Eastern Respiratory Syndrome (MERS) virus in the spike (S) protein region. This is the protein, especially the S1 attachment subunit, that is the principal determinant of host range, pathogenesis via cell fusion and communicabiity between human subjects.
The specificity of S is responsible in large part for the widespread and rapid communicability of SARS-CoV-2, with relatively low virulence in most of those infected (mortality varies but much less than 10%); in contrast, the spread of MERS is more easily contained, but its virulence is much higher, estimated at 35%.
A virus with the high communicability of SARS-CoV-2 and the virulence of MERS would be potentially apocalyptic in its impact.
Against that prospect, below is an annotated breakpoint sequence map for the reference sequence of MERS, delineating each occurrence of CAGAC or CAGAT, or its equivalent on the “minus” template strand, GTCTG. There are many fewer instances of these sequences in the MERS genome than in SARS-CoV-2, and most do not occur in a comparable location between the two viral genomes.
First of all, as noted by Graham and Baric (2010), each of the independently transcribed mRNAs are preceded by an identical hexanucleotide ACGAAC, that serves as the transcriptional regulatiory sequence TRS). Each could theoretically serve as a breakpoint bracketing any of the ORFs from S onward.
Second, there is a breakpoint sequence in MERS S, 21564CAGAC that is very close to the relative postion of 21691CAGAT in SARS-CoV-2. Homologous recombination at this point would not be likely to have a significant effect on protein structure.
Third, skipping one for the moment, there is a breakpoint sequence in MERS S, 25215CAGAT, that is similarly very close in relative position to 25047CAGAT in SARS-CoV-2.
Most importantly, there is a fourth breakpoint sequence, between the receptor binding domain of MERS and the S1/S2 junction, that is precisely conserved in sequence and position with the corresponding location of SARS-CoV-2. This breakpoint, 23577CAGAC in MERS and 23300CAGAC in SARS-CoV-2 (double-underlined in the pdf) is identical in both viruses at the same relative position in the S1 protein sequence, where it defines an identical dipeptide, QT.
There is potential, therefore, based on closely apposed or identical breakpoint sequences, that the bulk of the S1A and S1B domains of the S1 attachemnt subunit could be exchanged between MERS and SARS-CoV-2 in any mixed infection. While there is no indication that this has occurred in the wild, during the SARS-CoV-2 pandemic we face the unique situation of SARS-CoV-2 being present simultaneously in a substantial number of human beings worldwide. Any simultaneous outbreak of MERS, within or without those areas where is has been previously found prevalent, could produce the kind of mixed infection in humans that we know has resulted in frequent recombination among coronaviruses in the wild or in captive populations of animals. To the viruses, there is no known theoretical difference.
Public health authorities should especially guard against simultaneous spread of more than one coronavirus in the human population at the same time and in the same locations.