Despite intense efforts to sequence and analyze SARS-CoV-2 isolates, our understanding of the virus’s provenance is limited by incomplete genomic characterization of the Coronaviridae (CoV) family.
Serratus (https://github.com/ababaian/serratus) is an Open Science project for discovery of new virus sequences on an unprecedented scale. Serratus can search well over a million sequencing libraries per week for known and novel viruses, including RNA-seq, meta-genomic, meta-transcriptomic and environmental NGS datasets.
Here we report results from a preliminary survey of 1.14 million sequence libraries (26.78 petabases) from the NCBI Short Read Archive (SRA). We have uncovered previously unreported CoV species and identified thousands of CoV-positive libraries.
To facilitate rapid analysis of this data we are developing an R package, Tantalus (https://github.com/serratus-bio/tantalus), to interface with Serratus data. This project is under active development and we are seeking to establish immediate collaborations for analysis over the next several weeks.
Uncovering novel CoV species
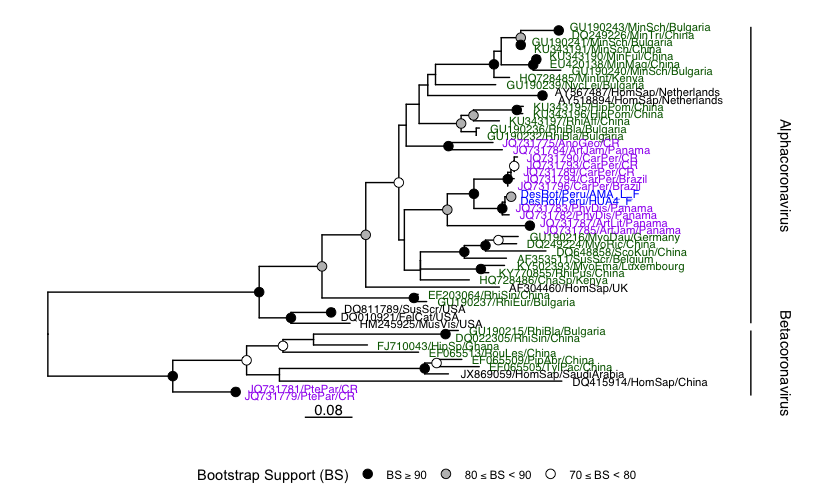
For just one example, in a Peruvian vampire bat (Desmodus rotundus) we identified a putative Alphoacoronavirus, via a partial sequence match at 93.4% identity to the RdRP of Bat coronavirus Trinidad/1CO7BA/2007 (GenBank accession: EU769558).
Assembly of this sequencing library with coronaSPAdes yielded a complete 29,264 nt viral genome. This is a new species of coronavirus based on RdRP, nucleprotein, membrane protein and replicase 1a, classified as a putative Alphacoronavirus falling outside all named sub-genera available in public nucleotide databases.
Alphacoronavirus identification case study
The author of the dataset had previously reported this virus as "DesRot/Peru/AMA_L_F” (Bergner et al. 2019 and see below, but it’s sequence is unpublished and was not in our query.
This is a clear proof of concept that there exists novel CoV within the Serratus data, there are likely scores more of these cases which we are actively working to uncover. Novel viruses from other family can also be identified with this workflow.
Join the Serratus Collaboration
Our primary objective is to accelerate global coronavirus research and assist in diagnostic and vaccine development with rich evolutionary CoV sequence data. All raw and processed Serratus data is freely and immediately available including a per-accession virus report of all vertebrate virus matches.
We are actively looking for collaborators for the intensive analysis of this data over the next four weeks. Expertise sought includes but is not limited to:
- Computational virology
- Phylogenetics and tree building
- Viral ecology and zoonosis modeling
- Database and web-interface development
- R package development
- AWS cloud computing
Researchers with access to large amounts of sequencing data from bats, wild rodents, or any sample taken from an animal with respiratory/GI disease are also sought. We are offering to generate a no-cost virus-report within 24 hours on upto 250,000 libraries. We only ask that adequate meta-data is provided and if samples are CoV+, the reports and CoV viral assemblies be shared immediately and without restriction.
Computational architecture
In February 2020, AWS mirrored the NCBI Short Read Archive (SRA) onto their S3 servers as an Open Data-set which allows for an unprecedented rate of access to raw sequencing data.
To perform the ultra-high throughput CoV search, we employed AWS cloud HPC with a 22,500 vCPU cluster (1460 x R5.xlarge, 4120 x C5.xlarge, and 90 x C5.large EC2 instances). Using this hyper-parallelized architecture we could bypass conventional networking and disk IO limitations to achieve a processing rate exceeding 500,000 sequencing libraries per day at a cost of ~$0.01 per library.
Our viral search query is a CoV pan-genome composed of all CoV Genbank sequences clustered at 99% identity and all non-retroviral “representative virus sequences” in RefSeq. We employ bowtie2 as the aligner which can detect short-read sequences at up to 20% nucleotide divergence. These alignment files are then summarized into a report file for import into R and downstream processing.
Conclusion
Serratus is an open science project; we are actively seeking to establish collaborations starting immediately to translate these data into a meaningful community resource in the fight against COVID-19. By expanding the known repertoire of coronaviruses together, we can not only help determine the origins of this pandemic, but help prevent another one.