Part 1 of this post can be found here:
https://pando.tools/t/selection-analysis-identifies-significant-mutational-changes-in-omicron-that-are-likely-to-influence-both-antibody-neutralization-and-spike-function-part-1-of-2/771
Omicron mutations at neutral or negatively selected S-gene sites might only be adaptive when they co-occur
Given both the apparent selective constraints on arising mutations at the cluster region 1, 2 and 3 sites in SARS-CoV-2 and other Sarbecoviruses, and the rarity of observed mutations at these sites among the millions of assembled SARS-CoV-2 genomes (despite evidence that individually such mutations do regularly occur during within-host evolution; Figure 3), it is very likely that Omicron mutations at cluster region, 1, 2 and 3 sites are maladaptive when present on their own. Nevertheless, the presence of mutations at these sites in Omicron, a virus that is clearly highly adapted, suggests that these mutations might interact with one another such that, when present together, they become adaptive. Therefore while individually the mutations might decrease the fitness of any genome in which they occur, collectively they might compensate for one-another’s deficits to yield a fitter virus genotype.
Positive epistasis of this type has, in fact, already been demonstrated between the cluster 2 mutation, S/Q498R, and the pivotal mutation of the 501Y SARS-CoV-2 lineages, S/N501Y. Whereas S/498R only marginally impacts the affinity of Spike for human ACE2 when present with S/501N (18), it increases ACE2 binding affinity approximately four-fold when present with S/501Y (19).
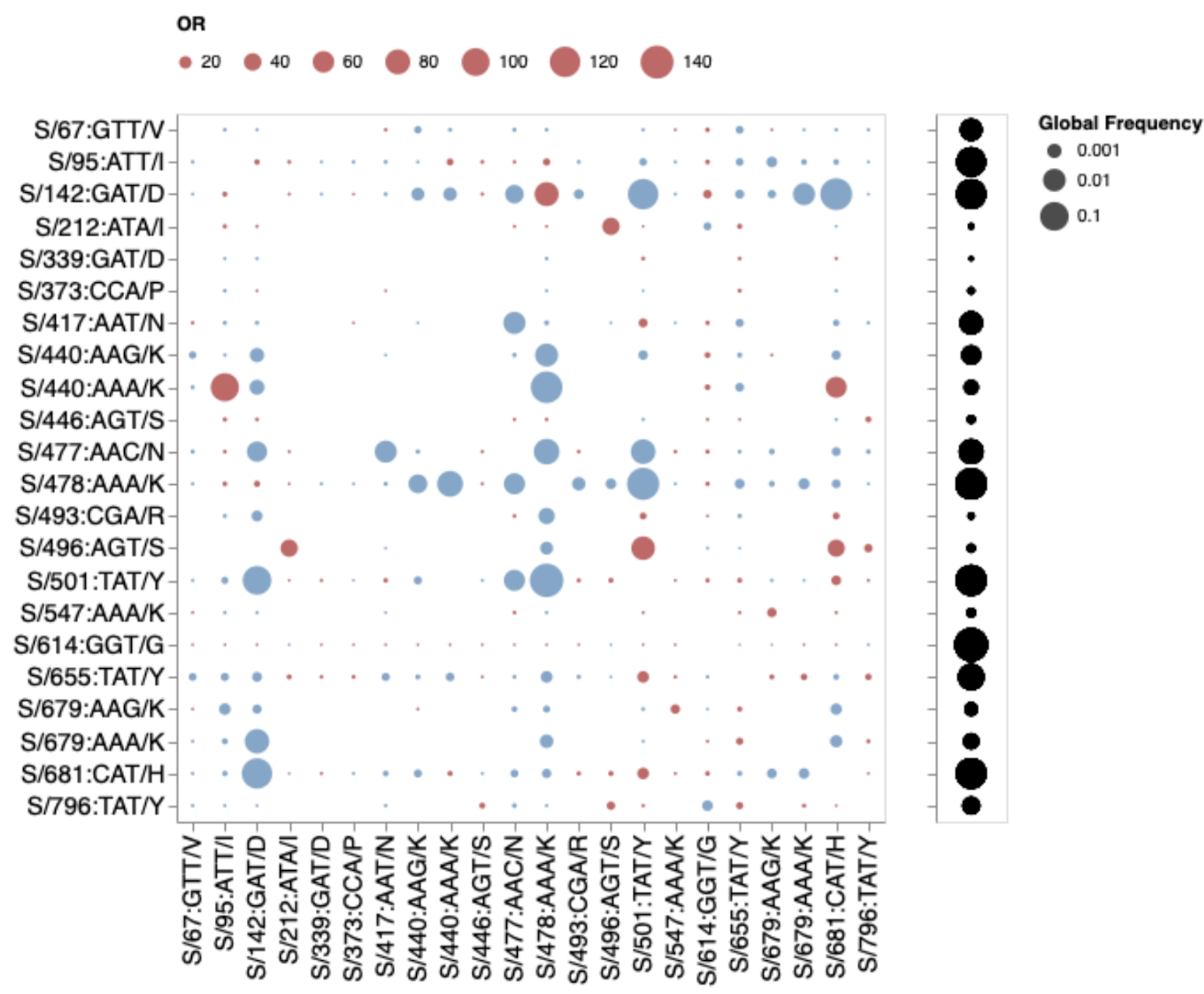
If mutations in the three cluster regions do epistatically interact with one another, then one might expect that selection would favour their co-occurrence either within individual SARS-CoV-2 genome sequences that have so far been sampled, or as minor variants within unassembled intra-patient sequence data. We failed to detect such associations in any systematic manner (Figure 6). While there are individual pairs of Omicron mutations that co-occur more frequently than expected by chance (e.g. 440K in the presence of T95I), they do not involve cluster 1, 2, and 3 mutations. Furthermore, many of the Omicron mutation pairs occur together less frequently than expected by chance (e.g. 478K and 501Y). Rather than reflecting an absence of epistasis between the cluster 1,2, and 3 mutation sites our failure to detect the co-occurrence of Omicrom mutation pairs at these sites simply reflects the rarity of these mutations within both assembled SARS-CoV-2 genome sequences and raw intra-patient sequence datasets (Figure 3).
Whether or not epistasis is extensively operating between mutations in the three cluster regions, the amino acid changes caused by these mutations in Omicron likely represent a substantial remodelling of two functionally important regions of Spike: the receptor binding domain and the fusion domain. If epistasis is operating between the mutations then it would simply mean that one might expect one or two major functionally important mutations in each of the clusters to have had adverse structural impacts on Spike that were compensated for by other mutations in the clusters.
Figure 6. Patterns of co-occurrence of Omicron amino-acid residues in circulating SARS-CoV-2 S-gene haplotypes from other lineages (data up to October 15, 2021). Only mutations occurring in at least 10 haplotypes are shown. All sequences having exactly the same S sequence count as a single unique haplotype; instead of counting raw sequence numbers, this approach focuses on the number of unique genetic backgrounds in which pairs of codons co-occur. Circles show odds ratios for finding the mutation on the X axis when the mutation on the Y axis is also present (vs when it is not present). Red circles depict OR > 1, while blue circles 1/OR for OR < 1. Black circles on the right show the fraction of globally sampled SARS-CoV-2 S-gene haplotypes which carry the corresponding mutation.
The cluster region 3 encoded amino acid changes in the part of Spike that is responsible for membrane fusion suggest that the membrane fusion machinery of the Omicron spike has been overhauled. The mutations in cluster regions 1 and 2 fall within the receptor binding domain (RBD) encoding part of the S-gene. These mutations, together with those at S/417, S/440, S/446 might be indicative of an extensive remodelling of the ACE2 receptor binding surface; possibly to accommodate a major change in the way that spike interacts with ACE2 and/or other host cell receptors.
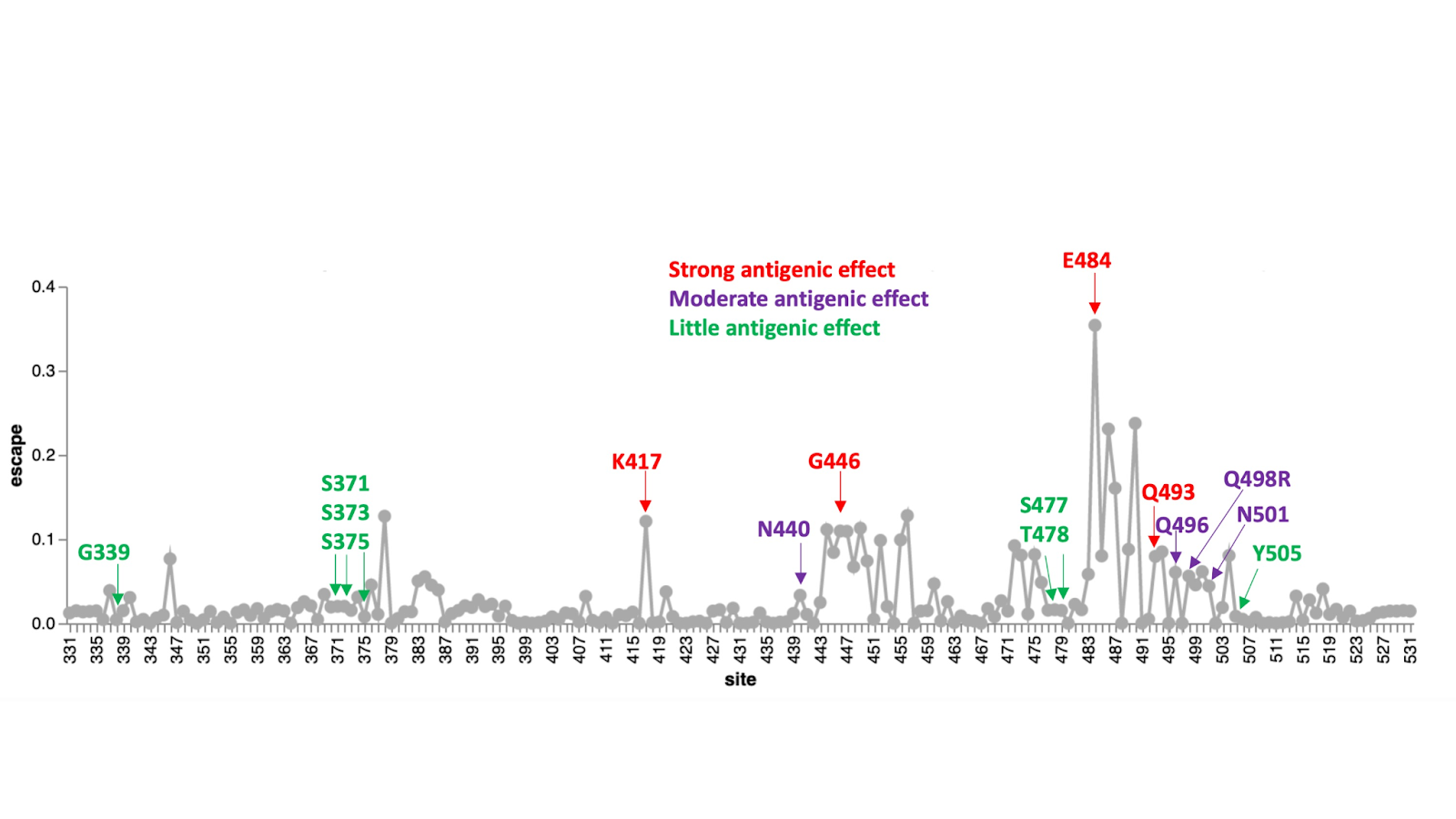
Of the cluster 2 sites that all fall within the receptor binding motif encoding part of the RBD, only S/498 and S/505 show signs of the Wuhan-Hu-1 encoded amino acid state having been selectively favoured in the past (S/498 in SARS-CoV-2 and S/505 in nCoV). No signs of any positive selection at the other cluster 2 sites in SARS-CoV-2 implies that changes at these and the negatively selected sites in cluster 2 have likely not individually contributed to effective immune evasion since the start of the pandemic. Deep mutational scans (Figure 7; (20)) have found little evidence that individual substitutions at S/505 have antigenic effects; S/496R and S/498R have only moderate antigenic effects, similar to those of the 501Y mutation. The exception that proves the rule that sites in this region might not be free to change in response to immune pressures is 493R. Given that 493R has a moderate antigenic effect, if it was not under selective constraints to sustain optimal degrees of ACE2 interaction (16) it should (but does not) display at least intermittently detectable signs of positive selection.
Figure 7. Experimentally measured effects of RBD mutations on binding of monoclonal antibodies at sites that differ between the Omicron variant and Wuhan-Hu-1. The line plot shows antibody binding escape measured by deep mutational scanning of the Wuhan-Hu-1 RBD (21), averaged across 36 monoclonal antibodies (8 class 1, 13 class 2, 7 class 3, and 8 class 4 antibodies). Sites that are mutated in the Omicron variant relative to Wuhan-Hu-1 are indicated and colored according to the predicted antigenic effect of mutations at that site (strong, moderate, or minimal). An interactive version of this plot is available at https://jbloomlab.github.io/SARS2_RBD_Ab_escape_maps/.
How and why have so many apparently maladaptive mutations been assembled within Omicron?
Given the manifest viability of Omicron there is a pressing need to understand how and why it accumulated so many mutations that, on their own at least, are apparently either selectively neutral or maladaptive. The sheer number of mutations in Spike and the genetic distance between Omicron and its nearest known SARS-CoV-2 relatives implies that the Omicron progenitor accumulated its unprecedented number of mutations during an extensive time period of undetected replication. When accurate molecular clock estimates are obtained of the time when Omicron last shared a common ancestor with other SARS-CoV-2 lineages, we will have an upper bound on the amount of time it took for Omicron to assemble its complement of mutations.
Omicron could have spent this period of intensive or prolonged evolution in a region that carries out minimal genomic surveillance or where access to, or utilization of, health care resources is low (the surveillance failure hypothesis). Alternatively, this viral evolution could have taken place within a long-term infection (or possibly serial long-term infections; the chronic infection hypothesis), or during spread within a non-human host population (the reverse-zoonosis hypothesis). Combinations of these evolutionary modes are also a possibility. We will only be able to distinguish between these hypotheses with more data. For example, if one or more SARS-CoV-2 lineages are discovered that are close relatives of Omicron then this would support the surveillance failure hypothesis, whereas if similarly divergent SARS-CoV-2 variants are discovered in either long-term human infections or in other animal species, these would support the other hypotheses.
Relative to evolution during normal SARS-CoV-2 person-to-person transmission, evolution within the context of either long-term infections or an alternative animal host could potentially have occurred at an accelerated pace (22,23). In these contexts purifying selection may have been relaxed somewhat relative to that occurring during normal human-to-human transmission: enough so for genomes carrying suboptimal combinations of epistatically interacting mutations to remain viable while fitter combinations were discovered via additional mutations and genetic recombination. In addition, chronic infections are not impacted by the tight transmission bottlenecks that can stochastically purge nascent adaptive mutations during normal transmission (24,25).
Sequential cycles of immune surveillance and viral immune escape within a long-term infection could also potentially explain the mutation clusters without the need to invoke compensatory epistatic interactions between mutations. Specifically, the clustered mutation patterns in the Omicron Spike are reminiscent of those seen in the HIV envelope protein as a consequence of sequentially acquired virus mutations that evade the progressively broadening neutralization potential of a maturing antibody lineage (26). While signs of negative selection at 9/13 of the mutated codons in the three cluster regions of Omicon are not entirely consistent with this hypothesis, the overwhelming contributor to these negative selection signals are the selective processes operating during normal short-term SARS-CoV-2 infections where the antibody-pathogen dynamics simply don’t have time to develop. It is possible that if purifying selection is relaxed at these sites during unusually prolonged infections, then neutralizing antibody evasion mutations might have been tolerable. Even if purifying selection were not relaxed, however, during a chronic infection the potential long-term fitness costs that are incurred by highly effective immune evasion mutations might frequently be offset by the immediate fitness benefits of evading neutralization.
Whatever the process that yielded the three clusters of rarely seen mutations in Omicron, now that it is being transmitted among people, any mildly deleterious immune evasion mutations it has accumulated might be substantially less tolerable. Likewise, some of the mutations it may have accumulated during its adaptation to transmission in an alternative animal species would now also potentially be somewhat maladaptive. If the rarely-seen mutations at negatively selected sites in the Omicron RBD that are known to be targeted by neutralizing antibodies begin reverting or acquiring clear second-site compensatory mutations over the coming months, it would best support the chronic-infection hypothesis in that such reversions would imply a trade-off between intra-host replicative and/or movement fitness and immune evasion. Alternatively, if reversion mutations occur at Omicron RBM sites that are known to impact human ACE2 binding but which have minor antigenic impacts, this would better support the reverse zoonosis hypothesis.
If, however, the rarely seen mutations in Omicron show no signs of reverting, rather than supporting one origin hypothesis over another, it would support the hypothesis that these mutations are broadly adaptive when they occur in the combinations found in Omicron. It would also indicate that rapid and substantial remodelling of important functional SARS-CoV-2 Spike domains is not just possible, but will likely recur in other lineages. The phenotypic impacts of such extensive genetic changes are very difficult to predict but, in the case of Omicron at least, the proximity of these changes to functionally important genome sites suggests the aspects of Spike function that are likely involved. The effects of the mutations in the three cluster regions on Omicron Spike function might be as similar to those caused by “normal” stepwise mutational changes as antigenic shifts are to antigenic drifts. Rather than just small tweaks in the antigenicity of Spike, its ACE2 binding properties or its membrane fusion functions, the clustered rarely seen mutations in Omicron’s RBD and fusion domain could cause quite big shifts in the way that Spike works.
Acknowledgements
We gratefully acknowledge all of the authors from the originating laboratories responsible for obtaining the specimens and the submitting laboratories where genetic sequence data were generated and shared via the GISAID Initiative, on which this research is based.