Selection analysis identifies significant mutational changes in Omicron that are likely to influence both antibody neutralization and Spike function (part 1 of 2)
Darren P Martin 1, Spyros Lytras 2, Alexander G Lucaci 3, Wolfang Maier 4, Björn Grüning 4, Stephen D Shank 3, Steven Weaver 3, Oscar A MacLean 2, Richard J Orton 2, Philippe Lemey 5, Maciej F Boni 6, Houriiyah Tegally7, Gordon Harkins 8, Cathrine Scheepers 9,10, Jinal N Bhiman 9,10, Josie Everatt 9, Daniel G Amoako 9, James Emmanuel San 7, Jennifer Giandhari 7, Alex Sigal 11, NGS-SA 12, Carolyn Williamson 13, Nei-yuan Hsiao 14, Anne von Gottberg 9,15, Arne De Klerk 1, Robert W Shafer 16, David L Robertson 2, Robert J Wilkinson 17,18,19, B Trevor Sewell 20, Richard Lessells 7, Anton Nekrutenko 21, Allison Greaney 22,23, Tyler Starr 22,24, Jesse Bloom 22,24, Ben Murrell 25, Eduan Wilkinson 7,26, Tulio de Oliveira 7,26, Sergei L Kosakovsky Pond 3
1 Institute of Infectious Diseases and Molecular Medicine, Division Of Computational Biology, Department of Integrative Biomedical Sciences, University of Cape Town, Cape Town 7701, South Africa
2 MRC-University of Glasgow Centre for Virus Research, University of Glasgow, Glasgow G61 1QH, UK
3 Institute for Genomics and Evolutionary Medicine, Department of Biology, Temple University, Philadelphia, PA 19122, USA
4 Bioinformatics Group, Department of Computer Science, University of Freiburg, Freiburg, Germany, usegalaxy.eu
5 Department of Microbiology, Immunology and Transplantation, Rega Institute, KU Leuven, Leuven, Belgium
6 Center for Infectious Disease Dynamics, Department of Biology, Pennsylvania State University, University Park, PA, USA
7 KwaZulu-Natal Research Innovation and Sequencing Platform (KRISP), School of Laboratory Medicine & Medical Sciences, University of KwaZulu- Natal, Durban, South Africa
8 South African National Bioinformatics Institute, University of the Western Cape, Cape Town, South Africa
9 National Institute for Communicable Diseases (NICD) of the National Health Laboratory Service (NHLS), Johannesburg, South Africa
10 SA MRC Antibody Immunity Research Unit, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
11 Africa Health Research Institute, Durban, South Africa
12 http://www.krisp.org.za/ngs-sa/ngs-sa_network_for_genomic_surveillance_south_africa/
13 Institute of Infectious Disease and Molecular Medicine, Department of Pathology, University of Cape Town, Cape Town, South Africa
14 Division of Medical Virology, University of Cape Town and National Health Laboratory Service, Cape Town South Africa
15 School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg
16 Division of Infectious Diseases, Department of medicine, Stanford university, Stanford, CA, USA
17 Wellcome Center for Infectious Diseases Research in Africa, Institute of Infectious Disease and Molecular Medicine and Department of Medicine, University of Cape Town, South Africa
18 Francis Crick Institute, Midland Road, London NW1 1AT, UK
19 Department of Infectious Diseases, Imperial College London, W12 0NN, UK
20 Structural Biology Research Unit, Department of Integrative Biomedical Sciences, Institute for Infectious Diseases and Molecular Medicine, University of Cape Town, South Africa
21 Department Of Biochemistry and Molecular Biology, The Pennsylvania State University, usegalaxy.org
22 Basic Sciences Division and Computational Biology Program, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA
23 Department of Genome Sciences & Medical Scientist Training Program, University of Washington, Seattle, WA 98195, USA3
24 Howard Hughes Medical Institute, Seattle, WA 98109, USA
25 Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Stockholm, Sweden
26 Centre for Epidemic Response and Innovation (CERI), School of Data Science, Stellenbosch University
The Omicron (B.1.1.529) SARS-CoV-2 variant of concern (VOC) identified in Southern Africa in late November 2021 is the product of extensive evolution within an infection context that has so far left no obvious traces of intermediate forms since it diverged from the B.1.1 lineage (presumably at some time in mid to late 2020). Three possible explanations for the missing intermediates are: (1) SARS-CoV-2 sampling in Southern Africa between May and September 2021 might have been too sparse or biased to detect low frequency variants amongst high numbers of Delta variant infections during this time period; (2) long-term evolution in one or more chronically infected people - similar to the proposed origin of lineages such as Alpha and C.1.2 (1) (2) (3) - where intermediate forms would have remained unsampled within the individual(s); and (3) a reverse zoonosis to a non-human host, followed by undetected spread therein, and a spillover back into humans. At present there is no direct evidence to support or reject any of these hypotheses on the origin of Omicron, but as new data are collected, its origin may be more precisely identified.
Regardless of the route that Omicron took to eventual community transmission, its genome accumulated 53 mutations relative to the Wuhan-Hu-1 reference strain, with 30 non-synonymous substitutions in the Spike encoding S gene alone. Here, we characterize the selective pressures that may have acted during the genesis of the Omicron variant and curate available phenotypic and genomic variation data associated with Omicron mutations. We are particularly interested in comparing evolutionary patterns at sites where Omicron has differences from the Wuhan-Hu-1 reference genome, most other SARS-CoV-2 lineages (including variation of SARS-CoV-2 in individual hosts), and closely related non-human sarbecoviruses. We use these comparisons to identify which Omicron mutations might contribute to transmission advantages, immune escape, or novel spike functionality. Our analysis identifies three clustered sets of mutations in the Spike protein, involving 13 amino acids that have previously been highly conserved across SARS-CoV-2 and other Sarbecoviruses. This dramatic about-face in evolutionary dynamics at these 13 sites suggests that Omicron’s Spike protein structure has accommodated significant sequence change, likely in response to selective pressures favoring increased transmission,immune evasion, or viral replication—either at the population level or in a single or group of chronically infected individuals—and has potentially acquired new functionality.
Many of the Omicron S-gene mutations are likely contributors to viral adaptation
Relative to the Wuhan-Hu-1 reference variant of SARS-CoV-2, Omicron has 30 non-synonymous substitutions in its S-gene. Sixteen of the codon sites where these mutations occur are presently, or have recently been, detectably evolving under positive selection when considering all SARS-CoV-2 genomic data prior to the discovery of Omicron (Table 1, Figure 1, Examining natural selection history on global SARS-CoV-2 genomes enabled by data from / Sergei Pond | Observable). For context, this fraction of positively selected sites (0.53) is approximately four times higher than the fraction of all SARS-CoV-2 S-gene sites that have ever shown any signals of positive selection (0.14).
Table 1. Frequencies in non-Omicron SARS-CoV-2 genomes of non-synonymous mutations seen in the S-gene of Omicron. Rows in bold indicate mutations at previously negatively selected or neutrally evolving sites. VOC columns track fold changes in mutation frequencies at corresponding sites in other VOC (before and after boundaries are defined to create somewhat balanced sizes of sequence sets; the boundary is 2021/04/15 for α,β,γ and 2021/06/01 for δ). If another amino-acid residue is included in parentheses, then this residue has increased in frequency at the same site. ↑ - 2-10x fold increase ↑↑ - >10x fold increase. ✓ - lineage defining/majority mutation. (*) in other human beta-CoV - consensus residue in species matches the Omicron residue; based on the sequence alignment from (4)

Figure 1. Selection signals that were evident at Omicron amino acid “mutation” sites in other SARS-CoV-2 lineages prior to the emergence of Omicron. All SARS-CoV-2 near full-length genome sequences present in GISAID (5) on 21 November 2021 that passed various quality control checks were split up into three month sampling windows and analysed using the FEL method restricted to internal tree branches (6) implemented in Hyphy 2.5 (7). This method was also used in (8). Red circles show sites under positive selection (selection favouring changes at amino acid states encoded at these sites). Blue circles show sites under negative selection (selection disfavoring non-synonymous changes). When no circle is shown, the corresponding site offered no statistical evidence for non-neutral evolution at a given time point. The areas of circles indicate the statistical strength of the selection signal (and not the actual strength of selection) within sequences sampled in the three months preceding the 1st day of the indicated months. Note that none of these analyses included any Omicron sequences, hence selection signals are derived solely from other SARS-CoV-2 lineages.

The observed substitutions at four of these sixteen sites (K417N, N501Y, H655Y, P681H) and a two-nucleotide deletion at one additional site (Δ69-70) are among the nineteen “501Y meta-signature” spike mutations that are likely highly adaptive within the context of 501Y lineage viruses such as the Alpha, Beta and Gamma VOCs (8) and, given that the Omicron mutations at these sites converge on those seen in these other VOCs, are likely to be adaptive in Omicron as well (sites coloured red in Figure 2).
A further four Omicron S-gene mutations are found in SARS-CoV-2 sequences belonging to other VOC lineages, and are either VOC lineage defining mutations (majority mutations), or are lower frequency mutations that have increased in frequency >2 fold between early and late VOC lineage circulation periods within sampled sequences belonging to these lineages (A67V in Alpha and Beta, T95I in Beta and Gamma, T478K in Beta, and N679K in Gamma; Frequency trends and selection detection of subsets of sites in SARS-CoV-2 genes / Sergei Pond | Observable and Table 1): an indication that these mutations too are likely adaptive in Omicron. Additionally, three other Omicron S-gene mutations either: (1) occur at the same codon sites as Alpha, Beta, Gamma or Delta lineage defining mutations but encode a different amino acid than these other lineages (E484A in Omicron and E484K in Alpha, Beta and Gamma); or (2) occur at the same codon sites as mutations in VOC lineages that increased in frequency > 2 fold between early and late VOC lineage circulation periods but encode a different amino acid than these other lineages (N440K in Omicron and N440S in Alpha; S477N in Omicron and S477I in Beta and Gamma). Lastly, the S/D796Y mutation occurs at one of the four sites identified as potential locations of adaptation in human beta-coronaviruses via the analysis of convergent evolutionary patterns and functional impact (Table 1) (4). All of these mutations likely have a substantial impact on the Omicron phenotype (coloured orange in Figure 2).
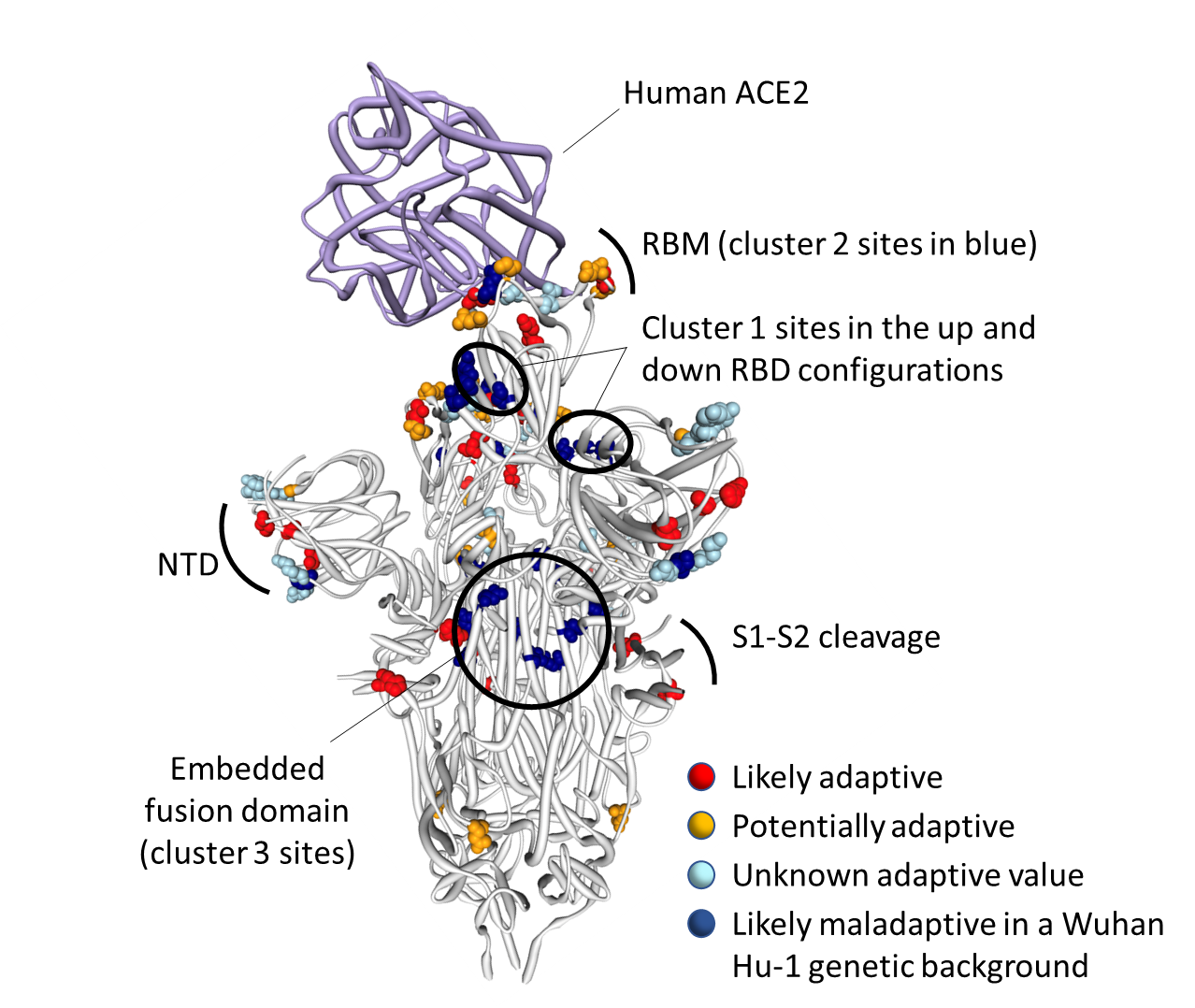
Figure 2. Distribution of Omicron amino acid replacements on the three dimensional SARS-CoV-2 Spike trimer. In this rendering of the trimer, one subunit is shown in the “up” or “open” configuration while interacting with human ACE2 (9). The other two subunits are in the “down” or “closed” configurations. Amino acids are color coded according to their likely contribution to viral adaptation in a Wuhan-Hu-1-like genetic background based on (1) patterns of synonymous and nonsynonymous substitutions at the codons encoding these amino acids in non-Omicron sequences, (2) patterns of mutational convergence between viruses in different VOCs and (3) increases in the frequency over time of VOC sub-lineages encoding amino acids that match those found in Omicron. NTD = N-terminal domain, RBD = Receptor binding domain; RBM = receptor binding motif. Locations of sites in the three clusters of Omicron mutations that are rarely seen and fall at either negatively selected (dark blue) and neutrally evolving (light blue) sites. An interactive version of this figure can be found here: SARS-CoV-2 ACE2/protein interaction and evolution for Omicron variant / Stephen Shank | Observable
Clusters of Omicron mutations occur at neutral or negatively selected S-gene sites
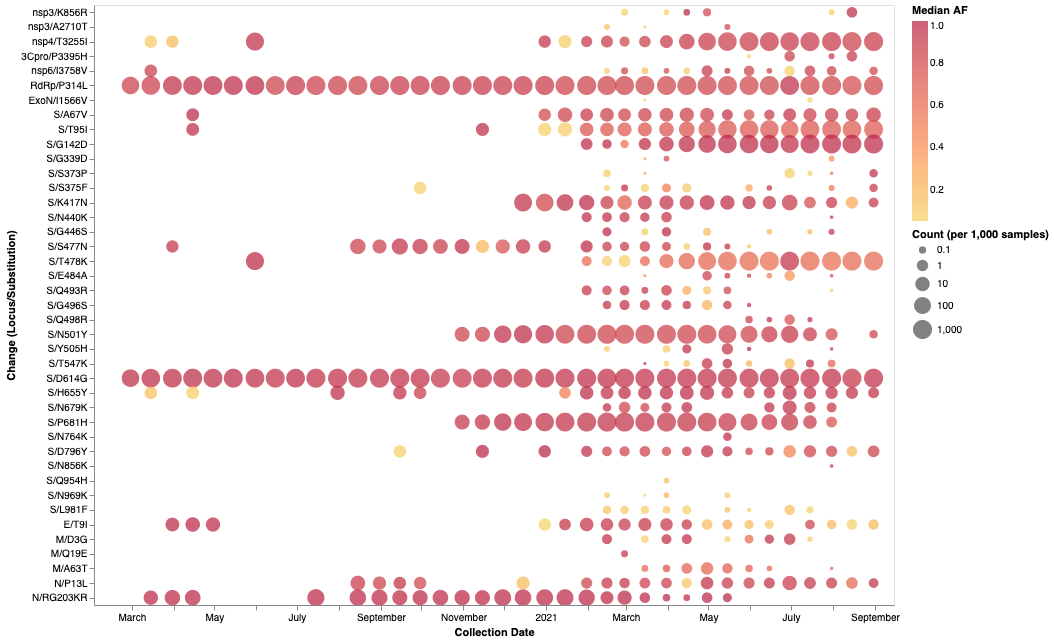
The mutations occurring at the 14 Omicron spike codons which display either evidence of negative selection or no evidence of selection (neutral evolution), have rarely been seen within previously sampled sequences (bold rows in Table 1; Omicron mutations in sequences up to Oct 2021 enabled by data from / Sergei Pond | Observable) indicating the action of strong purifying selection due to functional constraints. Despite the rarity of these mutations in assembled genomes, it is not uncommon to find them in within-patient sequence datasets (Figure 3), often at sub-consensus allelic frequencies. This indicates that, with the possible exceptions of S/N764K, S/N856K and S/Q954H, the mutations at these sites are not rare simply because they are unlikely to occur (note the sizes and numbers of dots corresponding to these mutations in Figure 3), but rather because whenever they do occur they are unlikely to either increase sufficiently in frequency to be transmitted (note the predominantly light orange/yellow colours of the dots corresponding to these mutations in Figure 3), or increase sufficiently in frequency among transmitting viruses to be detected by genomic surveillance.
Figure 3. Intra-host allelic variation seen at Omicron amino acid mutation sites in a subset of SARS-CoV-2 raw sequencing data since March 2020 analyzed using a standardized variant calling pipeline (10)… The areas of the circles indicate the proportions of raw sequence datasets (per 1,000 samples) where a mutation away from the Wuhan-Hu-1 consensus sequence was called. The colour of the circle indicates the median intra-patient allele frequency (AF) in datasets for which each mutation was detected. Mutations occurring at lower AFs are only present in a subpopulation of viruses in a particular host. The data has been generated by calling variants from read-level data of 230,506 samples from COG-UK, Estonia, Greece, Ireland, and South Africa: PRJEB37886, PRJEB42961 (and multiple other bioprojects with the study title: Whole genome sequencing of SARS-CoV-2 from Covid-19 patients from Estonia, PRJEB44141, PRJEB40277 and PRJNA636748. All variant calling data is available via ftp://xfer13.crg.eu/ and Global platform | COVID-19 analysis on usegalaxy.★).
On their own none of these 14 Omicron mutations at codon sites that have previously been evolving under negative selection prior to November 2021 would be expected to provide SARS-CoV-2 with any selective advantage. If the Omicron mutations observed at the ten negatively selected S-gene codon sites had occurred in the Wuhan-Hu-1 sequence, it is very likely that they would have been selected against. Specifically, since the start of the pandemic Spike proteins tended to function best whenever they had amino acids at these ten sites that were the same as those in the Spike encoded by the Wuhan-Hu-1 sequence.
It is clear that the amino acids encoded by 13 of the 14 mutated codon sites in the Omicron S-gene that either show evidence of negative selection or no evidence of any selection, cluster within three regions of the Spike three dimensional structure (light and dark blue sites in Figure 2):
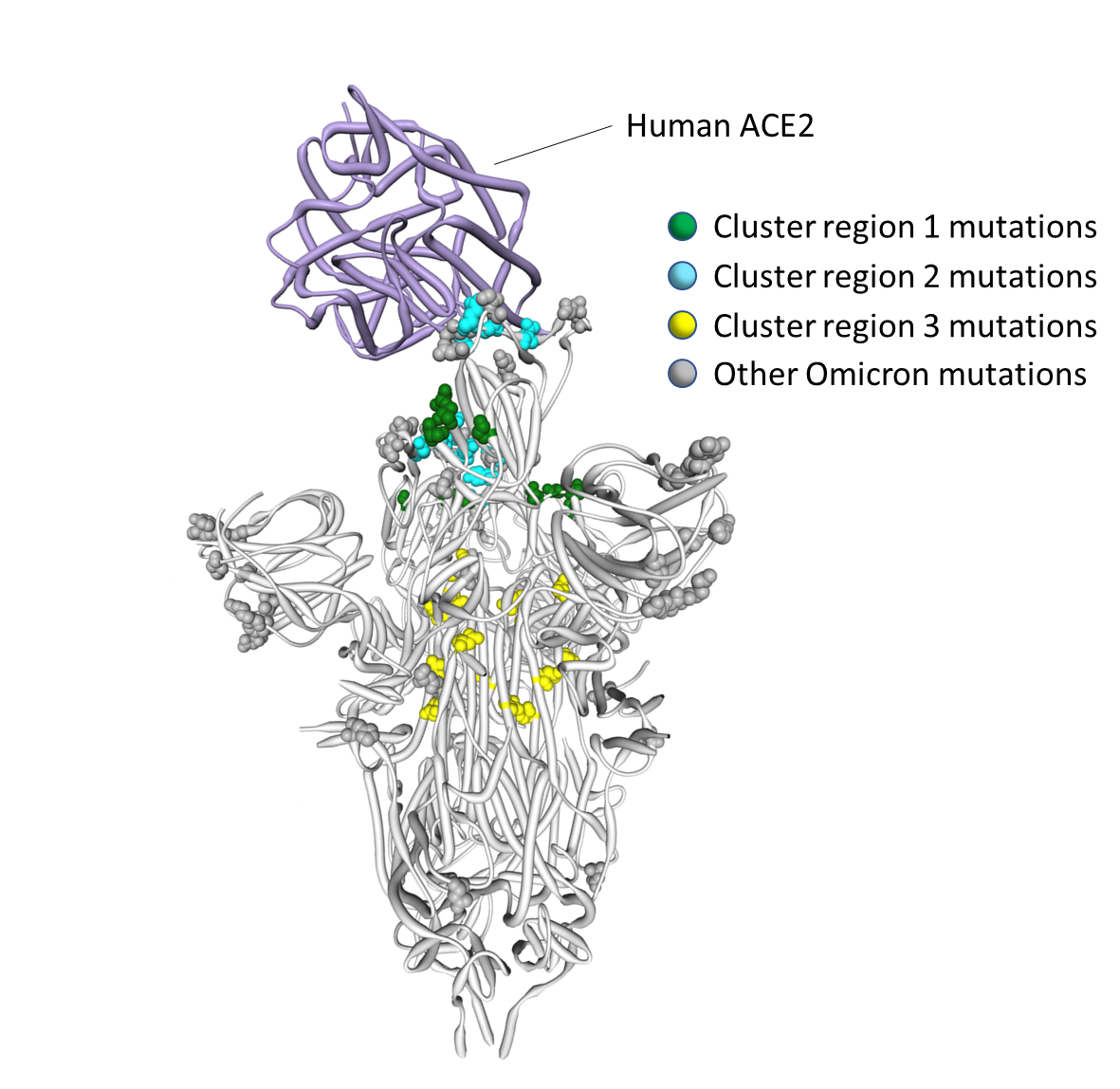
- Cluster region 1 in the RBD (green sites in Figure 4): codons/amino acids S/339, S/371, S/373 and S/375; may be targeted by some class 4 neutralizing antibodies (11).
- Cluster region 2 in the RBM (cyan sites in Figure 4) including codons/amino acids S/493, S/496, S/498, and S/505. This region is known to be targeted by class 1 and class 2 neutralizing antibodies. S/493 is, in fact, a known target of such antibodies. Accordingly S/Q493R (as occurs in Omicron) and S/Q493K escape mutations have been selected in VSV in vitro experiments (12) while the S/Q493K mutation has also arisen in the context of persistent SARS-CoV-2 infection (13).
- Cluster region 3 in the fusion domain (yellow sites in Figure 4): codons/amino acids S/764, S/856, S/954, S/969, S/981; a region of Spike not known to be currently targeted by neutralizing antibodies
Figure 4. Positions on the three dimensional SARS-CoV-2 Spike trimer of amino acids encoded by three clusters of Omicron codon sites that are evolving either neutrally or under negative selection in non-Omicron SARS-CoV-2 sequences. The Spike subunit interacting with human ACE2 is in the “up” configuration and the other two are in the “down” configuration (9). The cluster region 1 and 2 encoded amino acid changes in Omicron (in green and blue respectively) are within the receptor binding domain of Spike with the cluster 2 encoded changes located within the receptor binding motif. The cluster region 3 mutations are within the fusion domain of Spike. An interactive version of this figure can be found at SARS-CoV-2 ACE2/protein interaction and evolution for Omicron variant (clusters) / Stephen Shank | Observable
Selection patterns in Sarbecoviruses confirm that, on their own, many Omicron mutations would likely be deleterious
To determine whether patterns of selection at the Omicron-specific sites are broadly consistent with those occurring in the horseshoe bat-infecting SARS-related coronaviruses, in the Sarbecovirus subgenus to which SARS-CoV-2 belongs, we examined patterns of synonymous and non-synonymous substitutions in 167 publicly available Sarbecovirus genomes. Accounting for recombination, we tested for selection signatures at all 44 codons encoding amino acids that differ between Wuhan-Hu-1 and Omicron (Visualizing selection analysis results for evolution of nCOV (Nov 2021 update) / Sergei Pond | Observable). We specifically focused the analyses on selection signals in the subset of sarbecoviruses that are more closely related to SARS-CoV-2 in each recombination-free part of their genome: a group of sequences we refer to as the nCoV clade (14). Depending on the recombination-free genome region being considered, this clade was represented by between 15 and 27 sequences. We refer to the remaining sarbecoviruses as the non-nCoV sequences.
Of the 44 codon sites considered, 26 are detectably evolving under negative selection (FEL p-value <0.05; (7) (6) ) and one (S/417) under positive selection (MEME p-value <0.05; (15)) in the nCoV clade. This positive selection signal at S/417 reflects an encoded amino acid change from an ancestral V that is present in all background sequences, to a K that is specific to the nCoV clade. A K is also encoded at this site in Wuhan-Hu-1 but has since changed multiple times in various SARS-CoV-2 lineages: for example, to an N during the genesis of lineages such as Omicron and Beta and to a T during the genesis of the Gamma lineage.
We were, however, particularly interested in whether the cluster 1, 2 and 3 mutation sites in the S-gene were also evolving in a constrained manner (i.e., under negative selection) in the nCoV clade and, if so, what the selectively favoured encoded amino acid states were at these sites. Consistent with the hypothesis that the Wuhan-Hu-1 encoded amino acid states are generally constrained in the closest known SARS-CoV-2 relatives, the cluster 1 sites S/339, S/373 and S/375, the cluster 2 site S/505 and the cluster 3 sites S/764, S/856, S/969 and S/981 were all detectably evolving under negative selection in the nCoV clade viruses with the Wuhan-Hu-1 encoded amino acid state being favoured at all eight of the sites. Also consistent with the hypothesis, two of the remaining five sites across the clusters that were not detectably evolving under negative selection in the nCoV clade (S/371 and S/954) predominantly encoded the Wuhan-Hu-1 amino acid state in all sarbecoviruses. Only cluster 2 sites S/493, S/496 and S/498 seem to vary substantially across the Sarbecovirus subgenus.
What can the Sarbecoviruses tell us about the biological consequences of the rarely seen Omicron mutations?
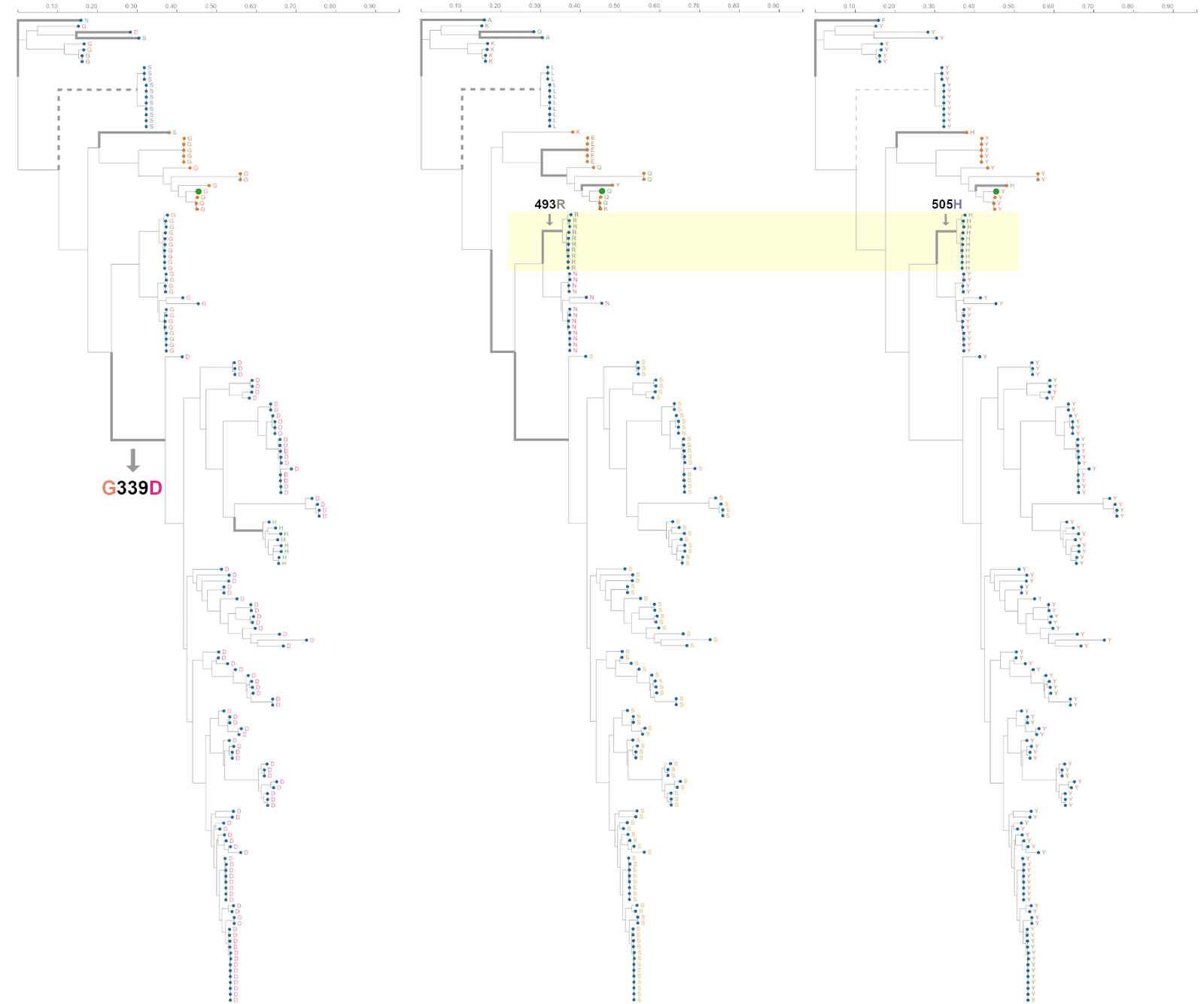
Despite the observation that, even among sarbecoviruses, Omicron mutations seen in cluster regions 1, 2 and 3 are only rarely seen, the instances where they do occur might be illuminating. For example, among the bat-infecting sarbecoviruses, the Omicron S/G339D substitution (in cluster region 1) has primarily to date been found among the bat-infecting viruses within a clade (Figure 5) that does not use ACE2 as a cell entry receptor (16). The change in receptor binding function in these viruses is, however, most likely due to two RBM deletions that are also specific to this clade. Further, cluster region 1 codon sites S/371, S/373 and S/375 encode a conserved serine (S) in almost all the analysed sarbecoviruses (164/167, 165/167 and 167/167 respectively). The change at sites S/371 and S/375 from an encoded polar residue (S) to a hydrophobic residue (an L at S/371 and an F at S/375) implies a substantial change in the biochemical properties of this region of Spike that has never before been seen in any sarbecovirus. These changes could be associated with SARS-CoV-2’s unique loss of N370 glycosylation site relative to all other sarbecoviruses (17), or packing of this surface with other Omicron changes in cluster 2 (e.g. S/Y505H) in the locked spike trimer structure.
As with SARS-CoV-2, the amino acids encoded at cluster region 2 sites (all of which fall within the RBM) vary substantially between different sarbecoviruses but without any associated signals of positive selection at these sites within the nCoV clade. Notably, the same Omicron encoded amino acids at codon S/493R and S/505H also co-occur in a clade of Sarbecoviruses that are closely related to SARS-CoV (virus accessions: KY417144, OK017858, KY417146, OK017852, OK017855, OK017853, OK017854, OK017856, OK017857); although S/493R (AY613951 and AY613948) and S/505H (MN996532, LC556375) can also occur independent of one another. Besides in Omicron, S/493R and S/505H are not found as a pair in any SARS-CoV-2 sequences. These mutations occurring along the same independent branch of the sarbecovirus tree (Figure 5) suggests that, rather than favouring changes at the sites individually, selection may favour simultaneous changes to S/493R and S/505H due to these residues together having a greater combined fitness benefit than the sum of their individual effects: a type of interaction between genome sites referred to as positive epistasis.
The region 3 cluster sites are conserved across the sarbecoviruses with almost all known viruses having the same residue as the Wuhan-Hu-1 SARS-CoV-2 strain. This supports the hypothesis that, when considered individually, the mutations seen at these fusion domain sites in Omicron are likely to be maladaptive.
Figure 5. Phylogenetic trees of 167 sarbecoviruses indicating patterns of selection at S-gene codons S/339, S/493 and S/505. Branches along which codons S/339 (left tree), S/493 (middle tree) and S505 (right tree) were inferred to be evolving under episodic positive selection (with the MEME method) are indicated with thick lines. The highlighted segments of the middle and right trees indicate that S/N493R and S/Y505H mutations occurred along . The trees represent evolutionary relationships between putatively non-recombinant sequence fragments in the genome region corresponding to Wuhan-Hu-1 Spike positions 324-654. Tree tips are annotated by amino acid states at sites 339, 493 and 505 (left to right). SARS-CoV-2 is annotated with a green tip symbol, the nCoV clade sequences with a tip symbol in orange and background sequences with a tip symbol in blue.
Part 2 of this post can be found here
https://pando.tools/t/selection-analysis-identifies-significant-mutational-changes-in-omicron-that-are-likely-to-influence-both-antibody-neutralization-and-spike-function-part-2-of-2/772