This is a continuation of a previous post.
We have reworked the clustering approach. We noticed a few k-mer distances could have been over-estimated due to sequencing differences at the genomes’ extremes. This did not affect the main results, as will be shown below. However, we decided to cluster the sequences again, this time from the the MSA, and repeat the analyses.
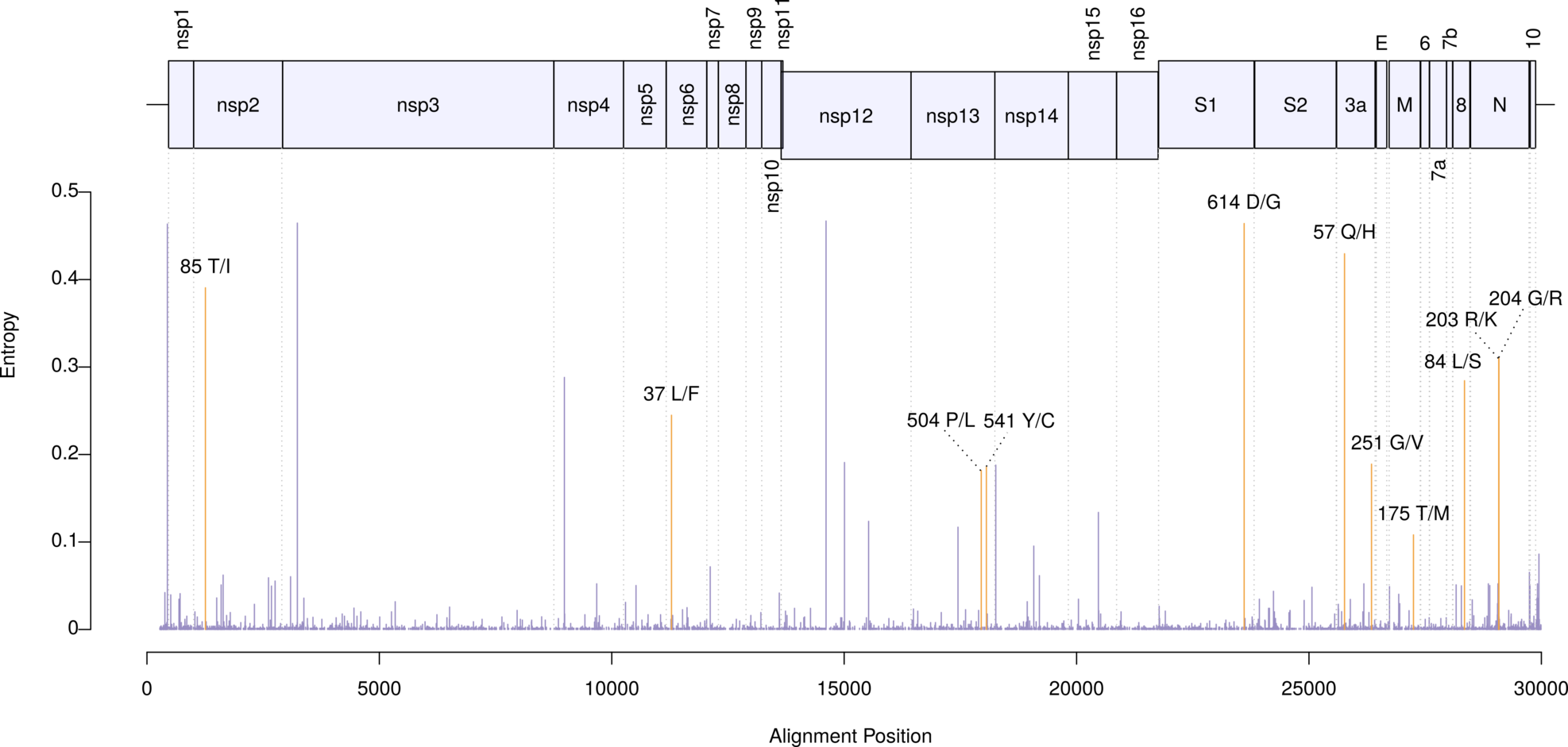
In addition, we performed a phylogeographic analysis of amino acid variants endorsed by at least 100 sequences. We found 11 such mutations. The least frequent variant (175M in protein M) was observed in 166 genomes. The figure below shows the polymorphisms locations along the genome. Entropies (y axis) were obtained from the RNA sequences. Orange-highlighted bars indicate the location of the nonsynonymous changes.
Phylogenetic structure analyses. The structure analyses gave results very close to the ones described in the previous post. As an example, the phylogenetic placement of the sequences from North America is shown below.

The sequences from North America preferentially clustered in tree sections A and E . Section A also contained many sequences from Europe (not shown).
Limitation in space was significant (p < 0.01) for Asia, Europe and North America.
Protein level phylogeographic analysis. To asses if the phylogenetic structuring process affected the virus proteins, we analyzed the geographic distribution and ancestral trajectories of the amino acidic polymorphisms described above. The amino acid variants distributions were very heterogeneous. Furthermore, ancestral amino acidic transitions were highly fitted to the virus phylogeny.
As an example, the figure below displays the distribution and ancestral trajectory of the 57Q/H polymorphism.
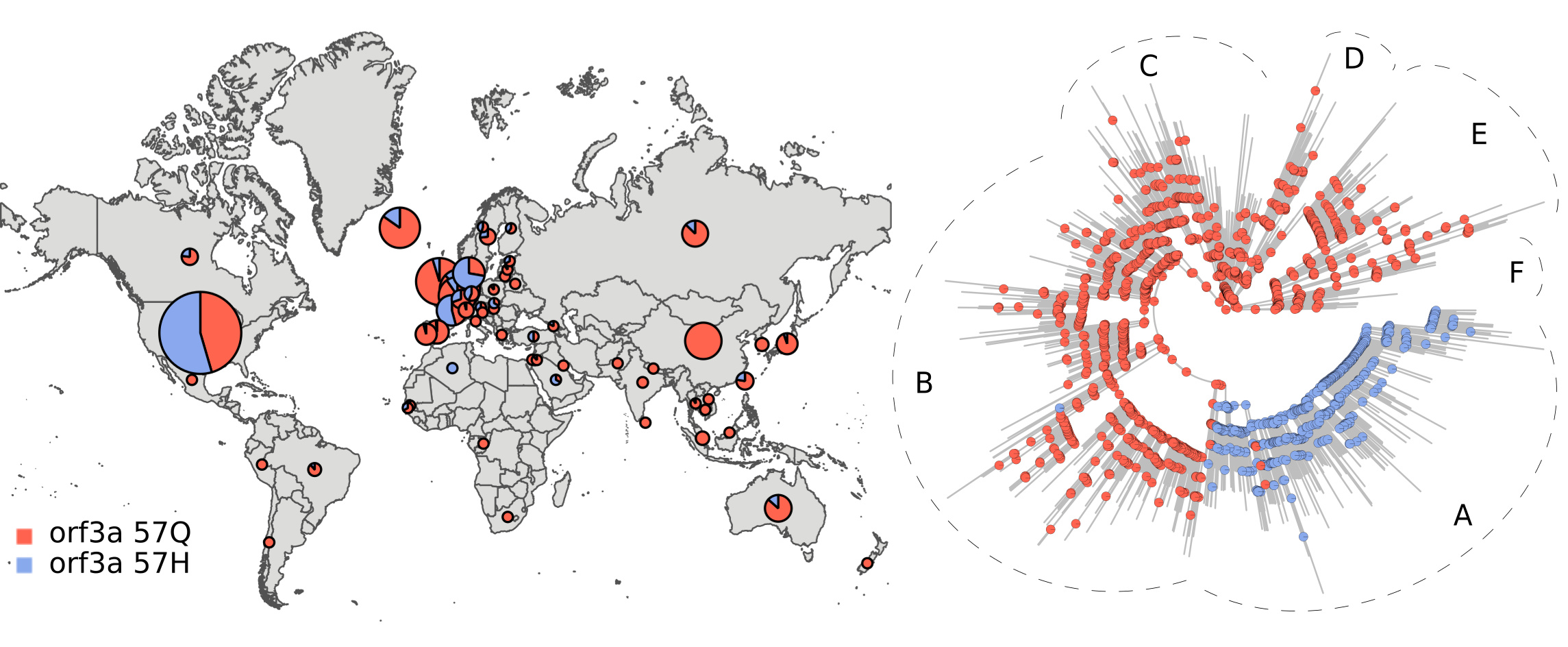
The large majority of ancestral nodes inside tree section A presented the 57H variant. This indicates that the divergence event leading to the split between tree section A and the rest of the phylogeny was likely accompanied by an orf3a protein mutation. The fact that the strains from tree section A were abundant in North America and Europe, suggests that dispersal to, or from, these regions was associated with 57Q/H polymorphism emergence.
A manuscript describing the full analyses is available here.