We report the first 11 SARS-CoV-2 genomes from Sierra Leone, showing evidence of multiple importations and community transmission in the country.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the etiologic agent of the current COVID-19 pandemic continues to pose a significant burden on public health. Sierra Leone (SL) reported its first COVID-19 confirmed case on the 30th March 2020 in the capital city; Freetown. Since then, a series of SARS-CoV-2 infections have been detected in all districts in Sierra Leone. Viral detection was performed using reverse-transcription polymerase chain reaction (RT-qPCR) in real-time, at both the Central public health reference laboratory (CPHRL) and Kenema Government Hospital (KGH) viral hemorrhagic fever (VHF) lab using the SARS-Cov-2 DaAn Gene kit. To better understand the virus in circulation at the molecular level, COVID-19 positive samples were enrolled for sequencing and molecular characterization at the KGH viral genome sequencing lab in Sierra Leone.
Sequencing was performed as follows: RNA was extracted using the QiAmp viral RNA mini kit (Qiagen) following manufacturers’ instructions. TIB Molbiol LightMix® SarbecoV E-gene kit was used to confirm all initial test results. We prepared unbiased metagenomics sequencing libraries using random primers and constructed libraries with Nextera XT. Sequencing libraries were pooled and sequenced using Illumina Miseq at KGH at a loading concentration of 12pM. We attempted reference based assembly on all samples using the viral-ngs pipeline version 2.1.0-rc8, and successfully assembled 11 full length genomes (>29,800 bp) of the virus with an appreciable mean coverage depth (Table 1). Here, we report the genomic epidemiology of human SARS-CoV-2 isolated from COVID-19 confirmed cases during the early period of the pandemic in Sierra Leone (March 30th - April 27th). To put these sequences in a regional and global context, we combined the 11 Sierra Leone sequences with the Nextstrain Africa build (auspice), downloaded on July 29th. The ML tree was constructed via IQ-tree (GTR substitution model) as included in the Nextstrain/Augur tools (Augur: A bioinformatics toolkit for phylogenetic analysis — Augur 21.1.0 documentation) and made available on DNAnexus as part of the viral-ngs workflows (viral-ngs: genomic analysis pipelines for viral sequencing — viral-ngs v2.1.33.15-70-g12567289 documentation).
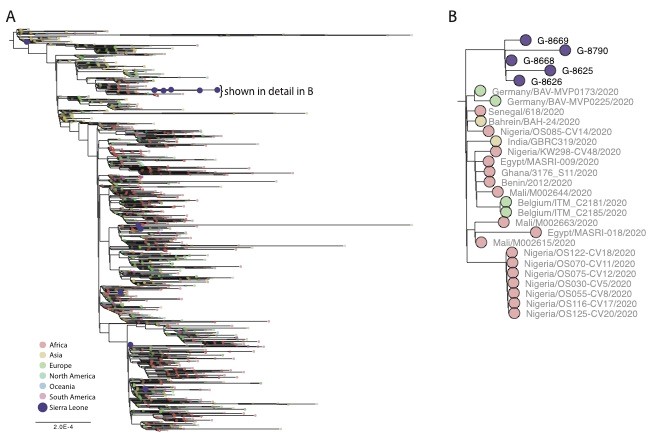
We observed that there were multiple introductions of the virus into SL as evidenced by the different clades (Clades 19B, 20B and 20C) of the virus in circulation (Figure 1A). This is consistent with the fact that at least 4 of the 11 patients reported recent international travel (Table 1), including the first confirmed cases of SARS-CoV-2 in Sierra Leone (G-8548). Additionally, we found evidence of community transmission as a subset of samples from SL clustered together on the tree (Figure 1B). Traditional epidemiology linked two of those samples (G-8625 and G-8626), a link that is supported by the genetics. Both the timing and number of mutations between other samples in this cluster suggest that these samples may be part of a larger, as-yet-unsampled clade. Further sequencing is needed to better resolve the extent of community spread in SL and to reconstruct transmission chains. We were also able to observe the presence of the derived (G) form of the GU280_gp D614G mutation in 5 of the 11 SL isolates sequenced to date (Table 1). We will continue to monitor this mutation and others in ongoing surveillance sequencing of cases from later samples in the outbreak in Sierra Leone.
SL_SARS-CoV-2 virological_Fig1.pdf (2.3 MB)
Figure 1. A. Maximum likelihood tree showing SL genomes (colored purple). B. Zoomed in a section of the ML tree showing 5 of the 11 Sierra Leone samples that cluster closely together, suggesting community transmission.
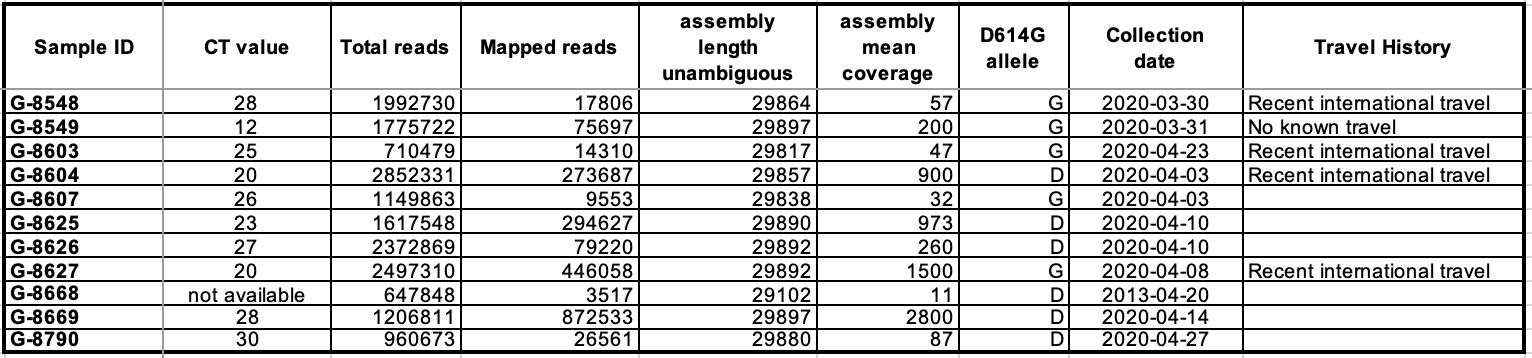
Table 1. Table reporting sequencing summary information, collection date and recent travel history for the 11 samples from which viral genomes were successfully assembled.
Data availability
Genome sequences are publicly available on GenBank in BioProject PRJNA655973, accession numbers;MT872492, MT872502 (Severe acute respiratory syndrome coronavirus 2 (ID 655973) - BioProject - NCBI)
Partners and Collaborators
Sierra Leone Ministry of Health and Sanitation
Kenema Government Hospital, Sierra Leone
Tulane University, Lassa fever research program, New Orleans, USA
African Centre of Excellence for Genomics of Infectious Diseases (ACEGID), Redeemer’s University, Ede, Nigeria
Sabeti Lab, Broad Institute of Harvard and MIT, Boston, USA
Andersen Lab, Scripps Research, USA
West African Emerging Infectious Diseases Research Centre
Funding
This work was supported by an award from Human health and heredity Africa (H3Africa) consortium (U54 HG007480) and the West African Emerging Infectious Diseases Research Centre (U01 AI151812).
Disclaimer and contact information
Please note that these analyses are based on work in progress and should be considered preliminary. Our analyses of this data are ongoing and a publication communicating our findings on these and other published genomes is in preparation. If you wish to use this data, please contact:
Prof. Sahr M. Gevao
Laboratory Pillar lead,
National COVID-19 Emergency Response Centre (NACOVERC)
Sierra Leone
Dr. Donald S. Grant (M D, MPH)
District Medical Officer-Kenema and Principal Investigator,
Lassa fever research program, Kenema Government Hospital,
Sierra Leone.
Email. [email protected]
Augustine Goba (BSc)
Director, Kenema Government Hospital Lassa fever (VHF) research Lab,
Kenema, Sierra Leone.
Email. [email protected]
John Demby Sandi (MSc.)
Laboratory research scientist. Lead, viral genome sequencing unit.
Kenema Government Hospital Lassa fever (VHF) research Lab,
Kenema, Sierra Leone.
Email. [email protected]
Twitter: @john_demby
Mambu Momoh (BSc.)
Laboratory research scientist,
Kenema Government Hospital Lassa fever (VHF) research Lab,
Kenema, Sierra Leone.
Email. [email protected]