SARS-CoV-2 diversity in Uganda, December 2020
MRC/UVRI & LSHTM Uganda Research Unit, Entebbe SARS-CoV-2 Sequencing Group, in close collaboration with Uganda Virus Research Institute and the Uganda Central Public Health Laboratory
Introduction
The novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) [1] and the associated disease Coronavirus Disease 2019 (COVID-19) [2][3] have now spread throughout the world, causing more than 81 million infections and over 1,700,000 death globally [4]. Sequence data from SARS-CoV-2 provide information on the transmission patterns and the evolution of the virus as it enters new regions. As COVID-19 vaccines become available, monitoring SARS-CoV-2 epitopes for changes is crucial. We have continued our efforts to monitor SARS-Cov-2 entry and movement through Uganda and have recently deposited an additional 113 SARS-CoV-2 genome sequences from Uganda in the GISAID database [5] [6] (GISAID accession numbers EPI_ISL_737931 to EPI_ISL_738043).
Overall status of the SARS-CoV-2 epidemic in Uganda
A summary of the current numbers of infections, mortalities and testing for SARS-CoV-2 in Uganda are shown in Figure 1. The slope of the cumulative cases line (Uganda plus Point of Entry (POE) cases Figure 1A) changed in July/August 2020 with a steeper increase due to elevated community cases. Since 1 Sep 2020, the cumulative community case number (Figure 1A dark blue) has exceeded the POE cases (Figure 1A light blue) with average daily positive cases rising from 27 in July 2020 to 235 in November 2020, to 479 for December 2020. The total SARS-CoV-2 positive deaths in Uganda is 248 (Figure 1B). Test positivity shows a steady increase and is now frequently above 10% (Figure 1D). Border entries from international flights were resumed on 1 October 2020, however the number of cases identified in return travellers and at POE has been modest (Figure 1A, light blue, grey markers) relative to the community cases (Figure 1A, dark blue markers) which are now dominating the Uganda epidemic.
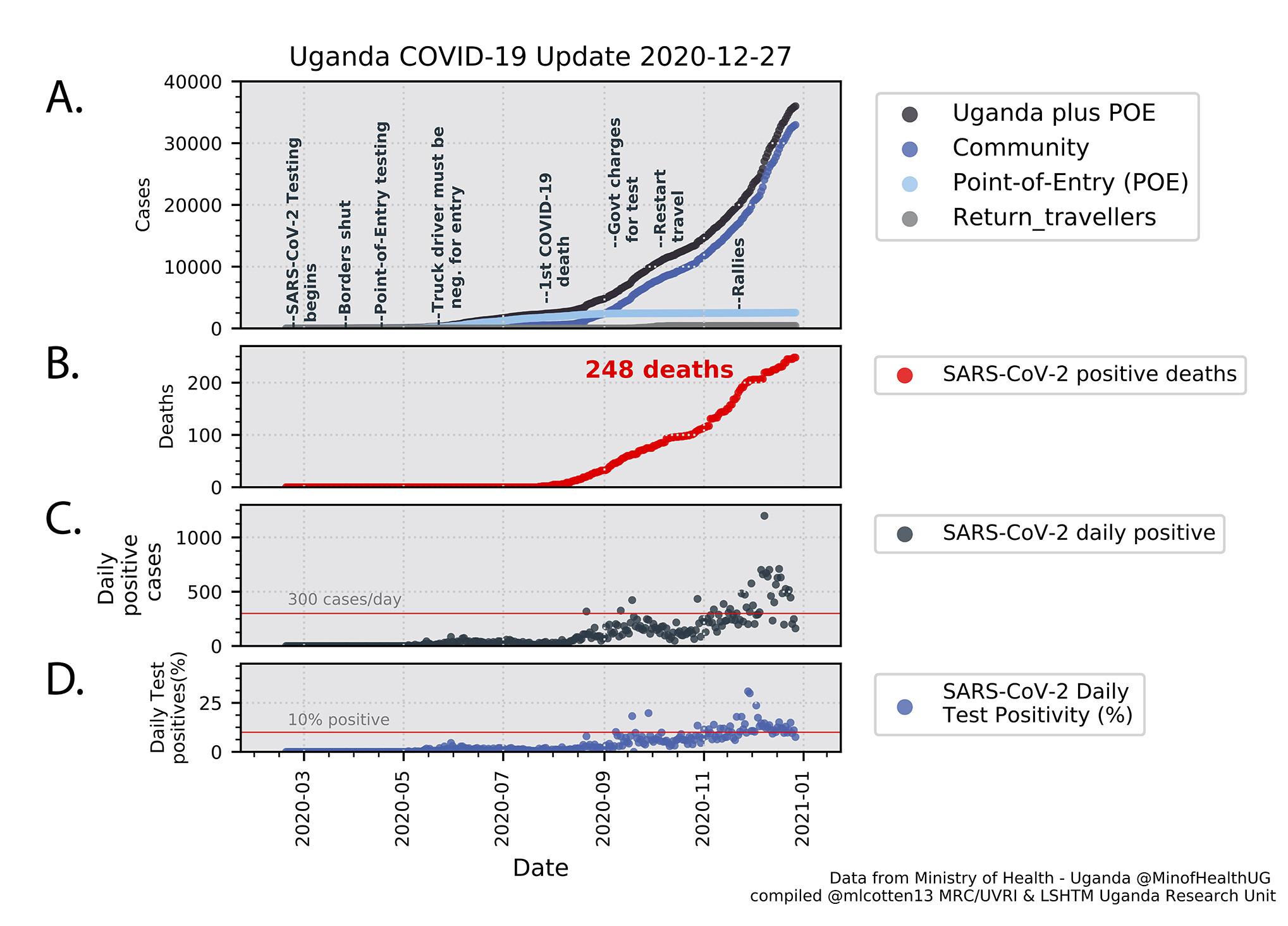
Figure 1. SARS-CoV-2 infections metrics. Panel A: Cumulative SARS-CoV-2 positive samples by case type. Total Uganda cases (black), Uganda community cases (dark blue), Point of Entry (POE) cases (light blue), Return travellers (grey). Important events in the epidemic are indicated. Pane B: Cumulative SARS-CoV-2 positive deaths. Panel C: Daily reported positive cases. Panel D. Daily test positivity (percentage of daily tests that were positive). Data were compiled from the Uganda Ministry of Health daily Twitter reports (@MinofHealthUG).
Virus sequence diversity including fatal cases
To monitor the epidemic, full genome SARS-CoV-2 sequences were generated from recent SARS-CoV-2 samples including 6 of the fatal COVID-19 cases in Uganda. A 1500bp-amplicon method, followed by MinION sequencing was used [7]. Assembled genomes were classified into the clades using Nextclade [8] and lineages using Pangolin [9][10]. Of the 131 Uganda full genomes now available, 50 (38%) belonged to lineage A (clade 19B) and the remainder belonged to a variety of B lineages with the majority lineages B.1 (N = 30; 23%) and B.1.5 (N = 17; 13%) found predominantly in truck drivers seeking to enter the country.
The genomes were used to construct a maximum-likelihood phylogenetic tree (Figure 2). Important details to note: the genome sequences from 6 lethal Ugandan cases were found in two lineages (A and B.1, Figure 2). Of note, the SARS-CoV-2 lineage A is far less prevalent than lineage B.1 in Europe, UK and USA; however, the presence of lineage A viruses from lethal cases and a large number of lineage A strains in community cases throughout the country suggested the lineage A is circulating in Uganda and capable of producing a severe infection. Additional B lineage genomes were initially observed in small numbers suggesting strain introduction limited spread in Uganda, similar to patterns observed in parts of the UK [11] and in Scotland [12]. Of interest for border control efforts in Uganda, viruses from truck drivers frequently appear basal to the observed community transmission clusters in the phylogenetic analysis (Figure 2), suggesting a possibly frequent source of virus entry into the country. Surprisingly, although a number of the minor B lineages entered Uganda with return travellers (Figure 2), the minor B strains have not contributed to community transmission in large numbers (compared to lineages A and B.1 and B.1.5).
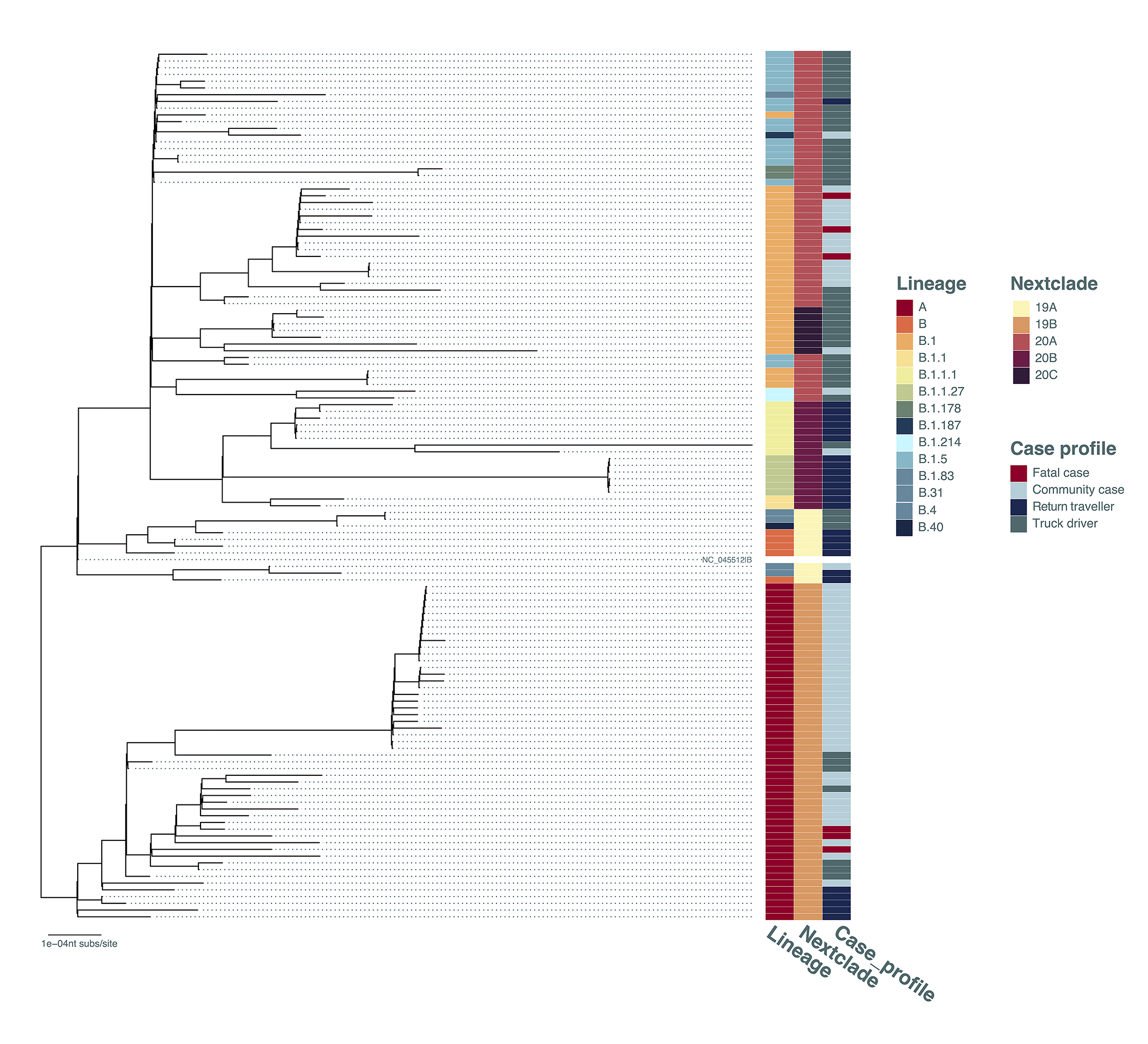
Figure 2. Maximum-likelihood phylogenetic tree comparing all currently available Uganda sequences (N=129, excluding several sequences with bad QC-scores in nextclade [8]). The tree was constructed under the GTRGAMMA model of evolution with 100 bootstraps in RAxML. The Pangolin lineages [10], nextclade clades [8] and case profile (community contact, truck driver, return traveller, fatal case) were shown. The tree was rooted where lineages A and B were split. The branch length is drawn to the scale of number of nucleotide substitutions per site.
Observed changes in spike protein
The spike protein is a critical target for COVID-19 vaccines, monitoring changes in this protein is essential as vaccines become available. The changes in spike protein observed in Uganda are documented in Figure 3. Most amino acid changes were single events with no apparent transmission observed (i.e. only singleton genomes were observed). However, several groups of changes were observed in more than one genome. The globally abundant D614G change was frequent, and a number of lineage A genomes with three persistent changes across the exposed S1 domain of spike (F157L, V367F and G613H) were observed. Two lineage B genomes with changes in the ACE2 binding region (L452R) were observed (Figure 3). Additionally, two examples of the two amino acid ∆H69V70 deletion, one in a lineage B lineage background and one in a lineage A background were observed (Figure 3). This spike deletion has been observed multiple times globally [13].
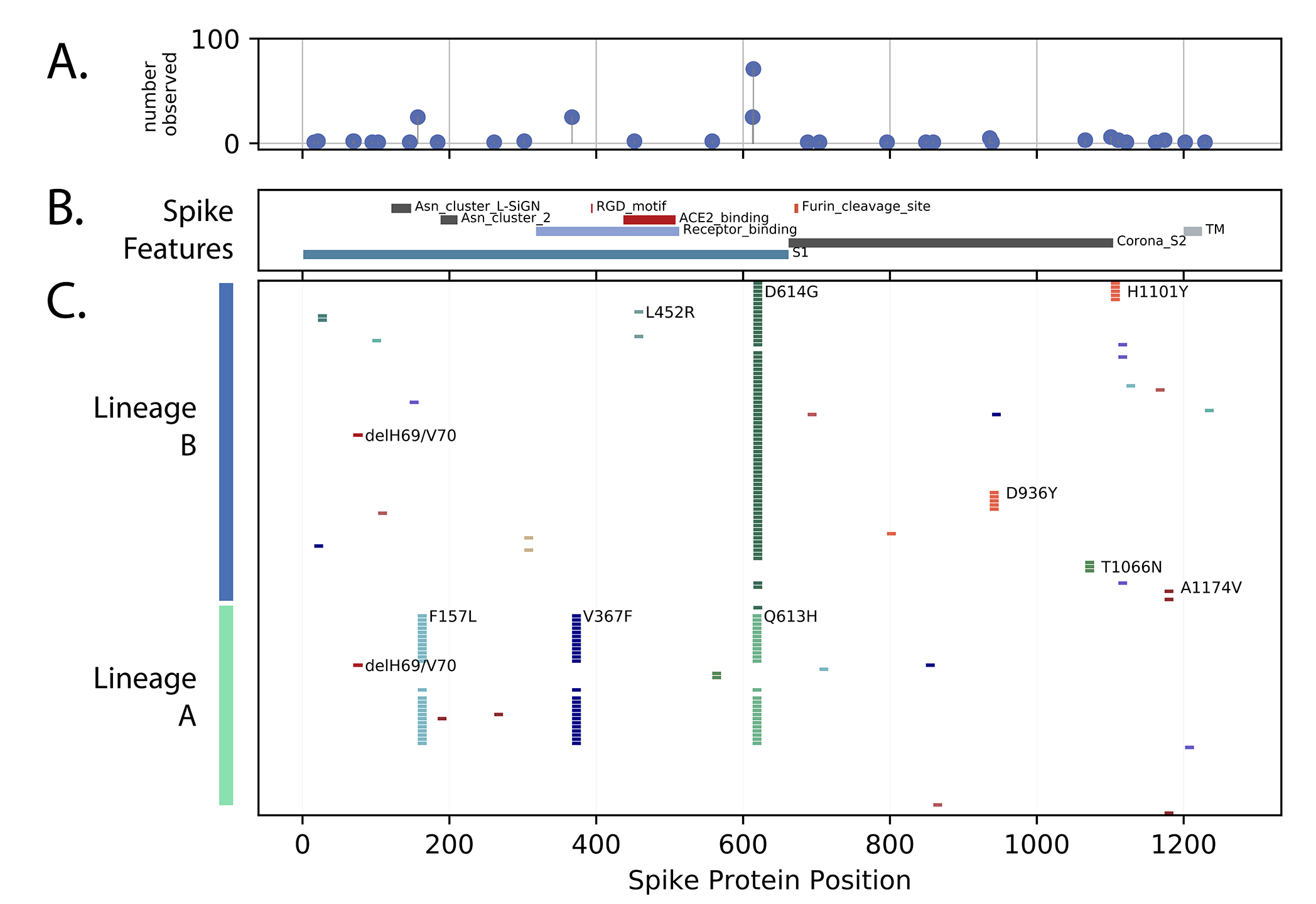
Figure 3. Spike protein changes observed in Uganda relative to the SARS-CoV-2 reference strain (NC_045512) encoded protein are documented. Panel C: Markers indication the positions of amino acid (AA) differences from the reference strain, changes observed in multiple genomes are annotated with the annotation (original AA position new AA). Panel B: The locations of important Spike protein features are indicated. Panel A: Compiled changes across the entire 131 genomes.
Conclusions
It is important to continue the surveillance of SARS-CoV-2 transmission and monitor the genomic diversity and evolution of the virus in Uganda, particularly as the vaccines are becoming available. Comments and questions on the new data and analysis are welcome.
Contributors
Pontiano Kaleebu1,2, Matthew Cotten1,3, Dan Lule Bugembe1, Deogratius Ssemwanga1, John Kayiwa2, Susan Nabadda4, Isaac Ssewanyana4, Patrick Semanda4
- MRC/UVRI & LSHTM Uganda Research Unit, Entebbe, Uganda
- Uganda Virus Research Institute, Entebbe, Uganda
- MRC-University of Glasgow Centre for Virus Research, Glasgow, UK
- Central Public Health Laboratories of the Republic of Uganda, Kampala, Uganda
Correspondence: Matthew Cotten ([email protected])
Acknowledgements
We thank all global SARS-CoV-2 sequencing groups for their open and rapid sharing of sequence data and GISAID for providing an effective platform for making these data available. We are grateful to the Oxford Nanopore Technologies and the ARTIC Network for their extensive support with protocols and analysis software. The SARS-CoV2 diagnostic and sequencing award is jointly funded by the UK Medical Research Council (MRC) and the UK Department for International Development (DFID) under the MRC/DFID Concordat agreement (grant agreement number NC_PC_19060) and is also part of the EDCTP2 programme supported by the European Union. The UMIC high performance computer was supported by MRC (grant number MC_EX_MR/L016273/1) to PK. The study is additionally funded by the Wellcome, DFID - Wellcome Epidemic Preparedness – Coronavirus (grant agreement number 220977/Z/20/Z) awarded to MC.
References
-
Edward C. Holmes, Yong-Zhen Zhang EC. Initial genome release of novel coronavirus Novel 2019 coronavirus genome. Virological.org [Internet]. 2020; . Available from: Novel 2019 coronavirus genome
-
Li Q, Guan X, Wu P, et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus–Infected Pneumonia. N Engl J Med. 2020; :NEJMoa2001316.
-
Yang X, Yu Y, Xu J, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. 2020; :S2213260020300795.
-
Lauren Gardner et al. Coronavirus COVID-19 Global Cases by Johns Hopkins CSSE. 2020; . Available from: ArcGIS Dashboards
-
Shu Y, McCauley J. GISAID: Global initiative on sharing all influenza data – from vision to reality. Eurosurveillance. 2017; 22(13):30494.
-
GISAID. The GISAID Initiative. 2020; . Available from: https://www.gisaid.org/
-
Cotten M, Bugembe DL, Kaleebu P, Phan MVT. Alternate primers for whole-genome SARS-CoV-2 sequencing [Internet]. Genomics; 2020 Oct. Available from: http://biorxiv.org/lookup/doi/10.1101/2020.10.12.335513
-
Trevor Bedford et al. Nextcladebeta v0.10.0 Clade assignment, mutation calling, and sequence quality checks. 2020; . Available from: https://clades.nextstrain.org/
-
Áine O’Toole, JT McCrone, Verity Hill and Andrew Rambaut. Pangolin COVID-19 Lineage Assigner. Available from: https://pangolin.cog-uk.io/
-
Rambaut A, Holmes EC, Hill V, et al. A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. Microbiology; 2020 Apr. Available from: A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology | bioRxiv
-
Page AJ, Mather AE, Le Viet T, et al. Large scale sequencing of SARS-CoV-2 genomes from one region allows detailed epidemiology and enables local outbreak management. Epidemiology; 2020 Sep. Available from: http://medrxiv.org/lookup/doi/10.1101/2020.09.28.20201475
-
Filipe ADS, Shepherd J, Williams T, et al. Genomic epidemiology of SARS-CoV-2 spread in Scotland highlights the role of European travel in COVID-19 emergence. Infectious Diseases (except HIV/AIDS); 2020 Jun. Available from: http://medrxiv.org/lookup/doi/10.1101/2020.06.08.20124834
-
Kemp S, Harvey W, Datir R, et al. Recurrent emergence and transmission of a SARS-CoV-2 Spike deletion ΔH69/V70. Microbiology; 2020 Dec. Available from: http://biorxiv.org/lookup/doi/10.1101/2020.12.14.422555