Response to “On the origin and continuing evolution of SARS-CoV-2”
Oscar A. MacLean*, Richard Orton, Joshua B. Singer, David L. Robertson.
MRC-University of Glasgow Centre for Virus Research (CVR).
*To whom correspondence should be addressed: [email protected].
Introduction
An analysis of genetic data from the ongoing COVID-19 outbreak was recently published in the journal National Science Review by Tang et al. (2020). Two of the key claims made by this paper appear to have been reached by misunderstanding and over-interpretation of the SARS-CoV-2 data, with an additional analysis suffering from methodological limitations.
The first claim
This criticism concerns the claim that there are two clearly definable “major types” of SARS-CoV2 in this outbreak and that they have differentiable transmission rates.
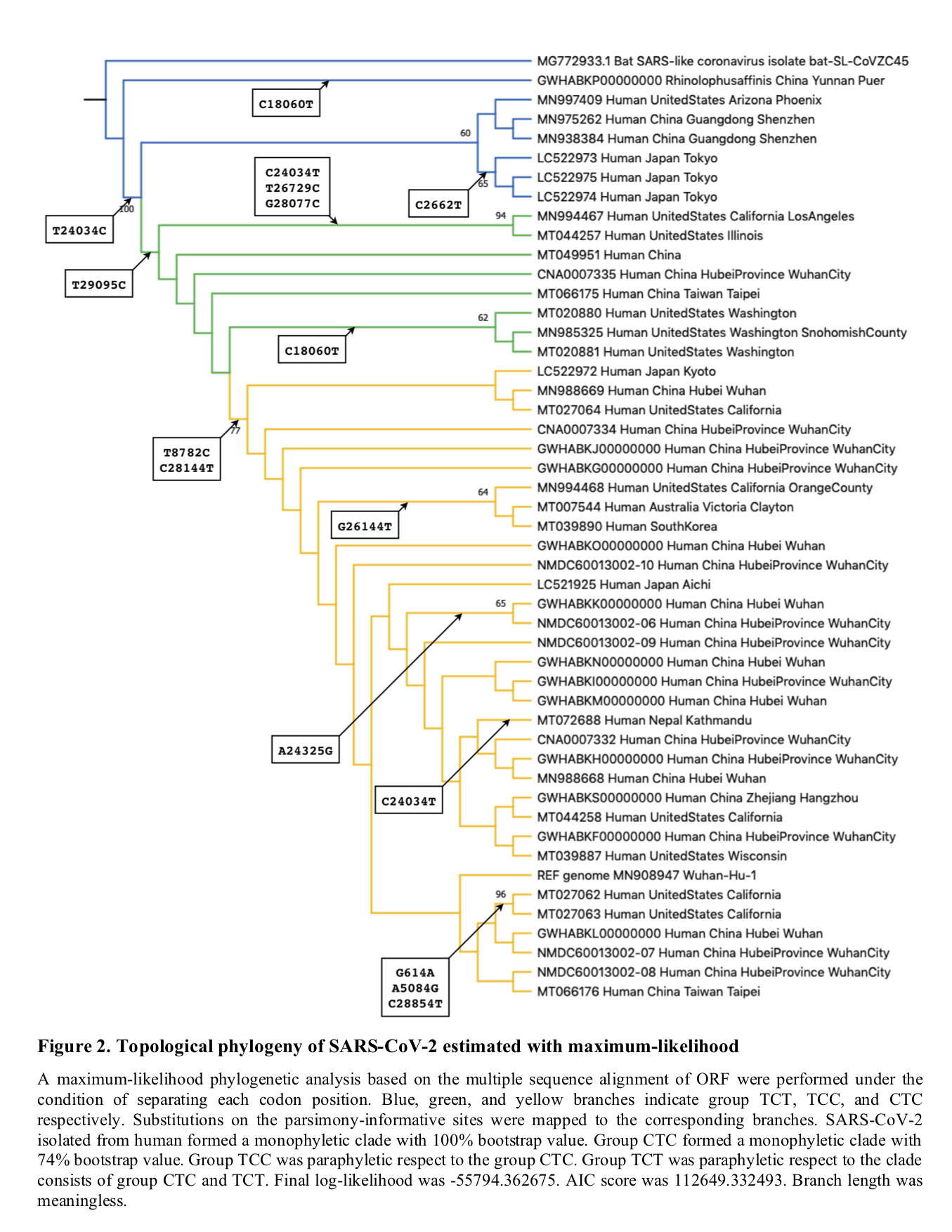
Tang et al. term these two types L and S type: “two major types (L and S types): the S type is ancestral, and the L type evolved from S type. Intriguingly, the S and L types can be clearly defined by just two tightly linked SNPs at positions 8,782 (orf1ab: T8517C, synonymous) and 28,144 (ORF8: C251T, S84L).”
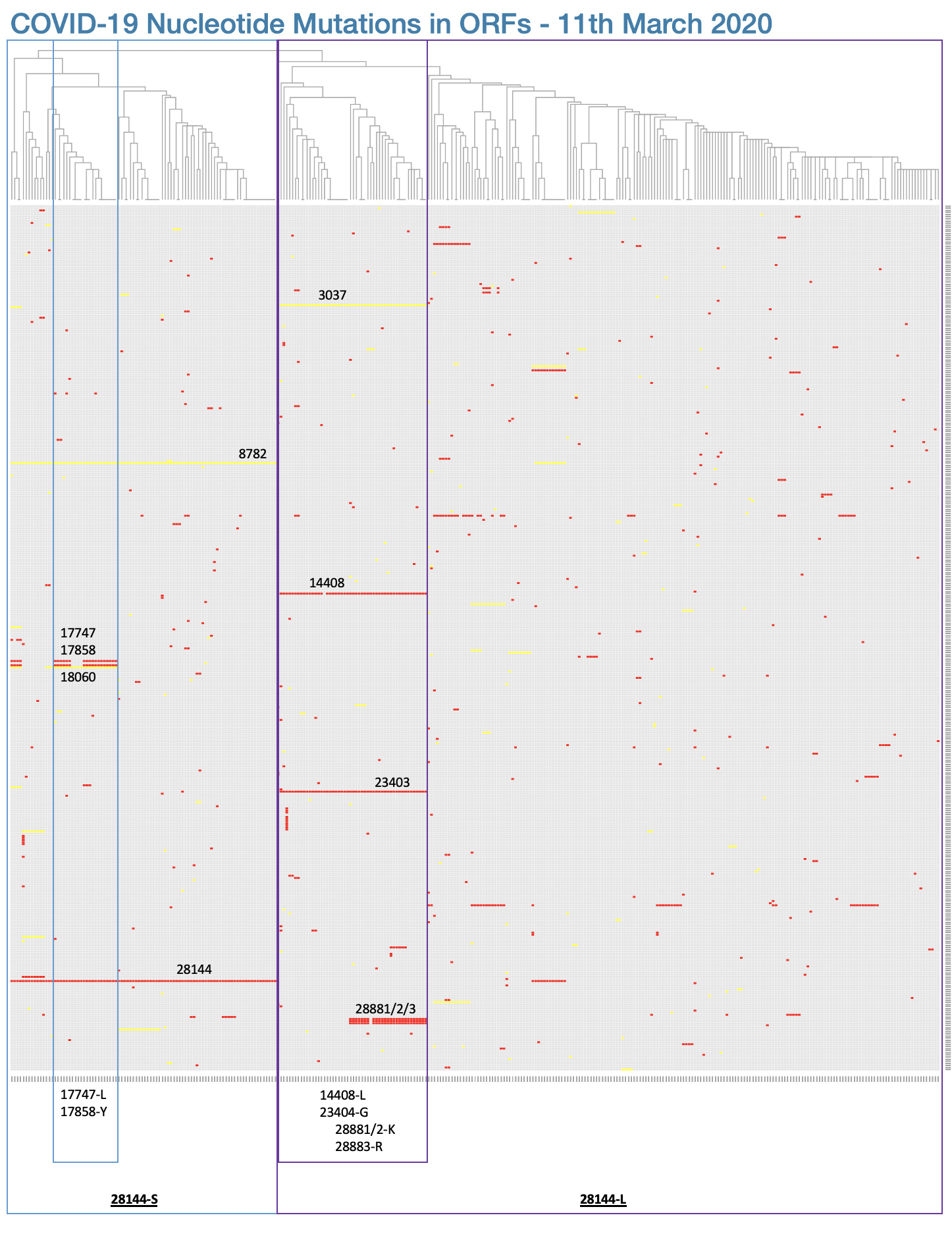
One nonsynonymous mutation, which has not been assessed for functional significance, is not sufficient to define a distinct “type” nor “major type”. As of 2nd March 2020, there are 111 nonsynonymous mutations that have been identified in the outbreak, these have been catalogued here in the CoV-GLUE resource and can be visualised in Figure 1. At current, there is no evidence that any of these 111 mutations have any significance in a functional context of within-host infections or transmission rates. Additionally, when you choose to define “types” purely on the basis of two mutations, it is not intriguing that these “types” then differ by those two mutations.
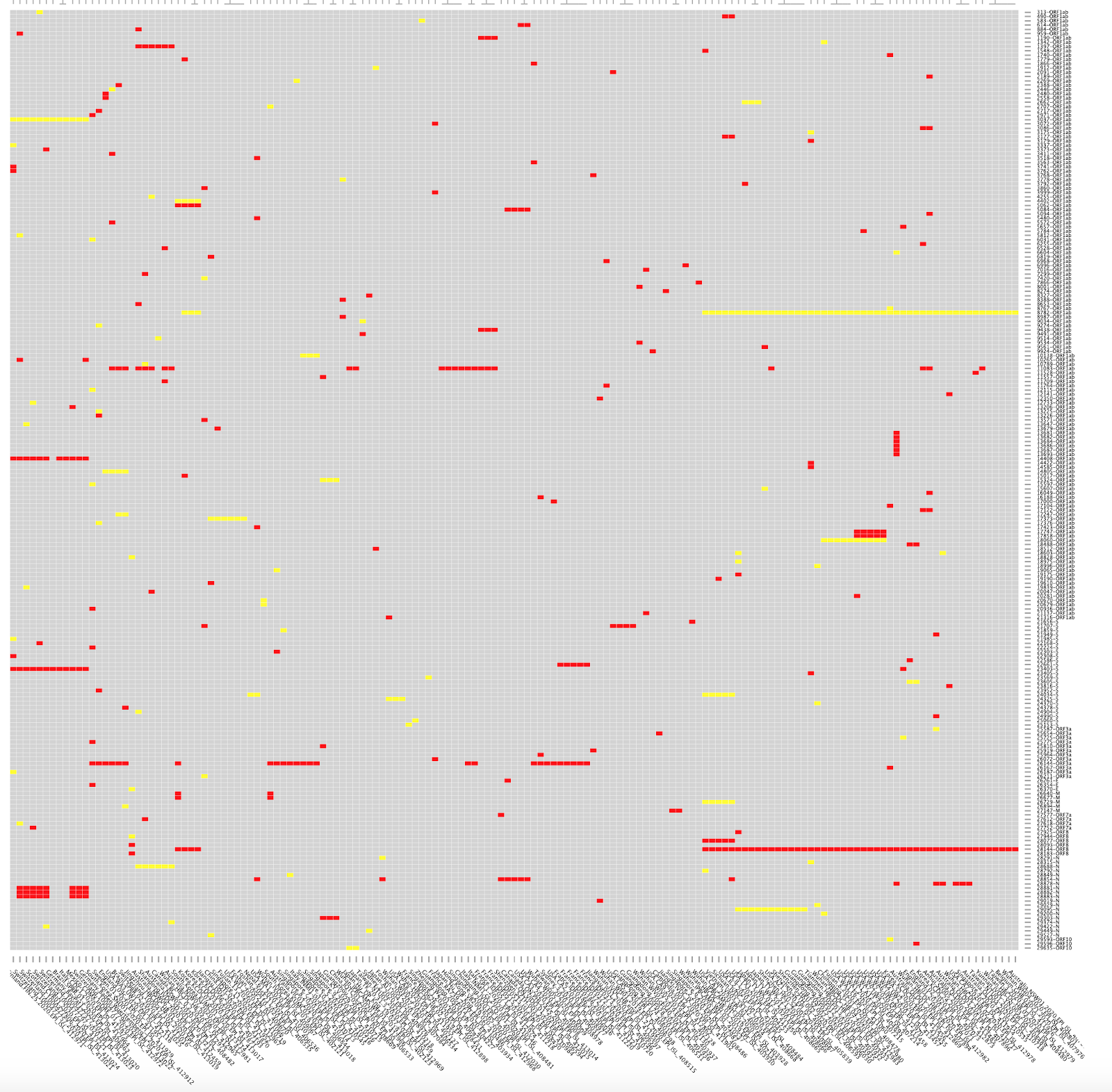
Figure 1. A visualisation of the 111 nonsynonymous mutations (red) observed to date in the COVID-19 outbreak by plotting a grid of mutations where each column is a sample and each row is one of the observed mutations in the phylogeny. The columns are ordered by the position of each sample in the phylogeny. Synonymous mutations are shown in yellow. The C251T (nonsynonymous) and T8517C (synonymous) mutations are visible on the right side of the plot.
However, they further claim that these two types have differing transmission rates: “Thus far, we found that, although the L type is derived from the S type, L (~70%) is more prevalent than S (~30%) among the sequenced SARS-CoV-2 genomes we examined. This pattern suggests that L has a higher transmission rate than the S type.” The abstract of the paper goes even further, stating outright that: “the S type, which is evolutionarily older and less aggressive…”
It is, however, important to appreciate that finding a majority of samples with a particular mutation is not evidence that viruses with that mutation transmit more readily. To make this claim would, at very minimum, require a comparison to be made to expectations under a null distribution assuming equal transmission rates. As this has not been performed by the authors, we believe there is insufficient evidence to make this suggestion, and therefore it is incorrect (and irresponsible) to state that there is any difference in transmission rates. Differences in the observed numbers of samples with and without this mutation are far more likely to be due to stochastic epidemiological effects.
Basic evolutionary theory predicts that selectively neutral mutations change in frequency over time through the process of genetic drift. In a viral outbreak, each transmission event from one infected person to another is a random probabilistic event, with some infected individuals transmitting more or less often than others. People may transmit at higher rates than others for a variety of reasons, e.g., because they cough onto their palms and use overcrowded public transport, or just because their friends and coworkers got lucky (or unlucky!). These small-scale epidemiological phenomena add up over time to create substantial variation in the frequencies of mutations observed during an outbreak.
Additionally, when a virus spreads to a new area/country that was previously uninfected, a founder effect can occur. As a small number of virus copies rapidly spread into an epidemic, any mutations in the initial viral infections will rapidly become very common, even if they were initially rare in the country that seeded the transmission. This is particularly likely to be the case in an outbreak caused by a novel virus such as COVID-19 as there are a large number of susceptible hosts for the virus. These founder effects have been observed in previous studies of viral outbreaks (e.g., Foley et al. 2004; Rai et al. 2010; Tsetsarkin et al. 2011). Combined, these factors mean that the frequency of a particular mutation in and of itself is not suggestive of any functional significance. Evidence from the widespread media uptake (35 articles at last count), and many comments on social media in response to this article, suggests that the unsupported claims made by Tang et al. have already spread undue fear.
It’s also important to appreciate that the smaller the population of viruses is, the more these small scale variations are likely to affect the frequency of mutations (in the same way that the more coins you flip, the closer to the 0.5 heads average you expect to be). Given that this mutation appears to have occurred very early on in the outbreak, when fewer individuals were infected, it’s frequency will very likely have been particularly influenced by genetic drift.
The second claim
Tang et al. compare the frequencies of nonsynonymous and synonymous mutations in the data, claiming that there is significant evidence of selection suppressing the frequency of nonsynonymous mutations in the outbreak. This analysis is flawed on three grounds:
(1) The numbers in this figure do not make sense. According to the presented data, seven (synonymous) mutations have a derived frequency of >50%, and two of these mutations have derived frequencies greater than 95% in the population. A cursory glance at the tree (Figure 2; taken from Nextstrain) shows that this cannot be true. “Derived” in this context should mean since the last common ancestor of the outbreak. For two mutations to have derived frequencies greater than 95%, there would need to be a small number of samples which branch as a sister lineage to the rest of the outbreak tree. However, this is not the case.
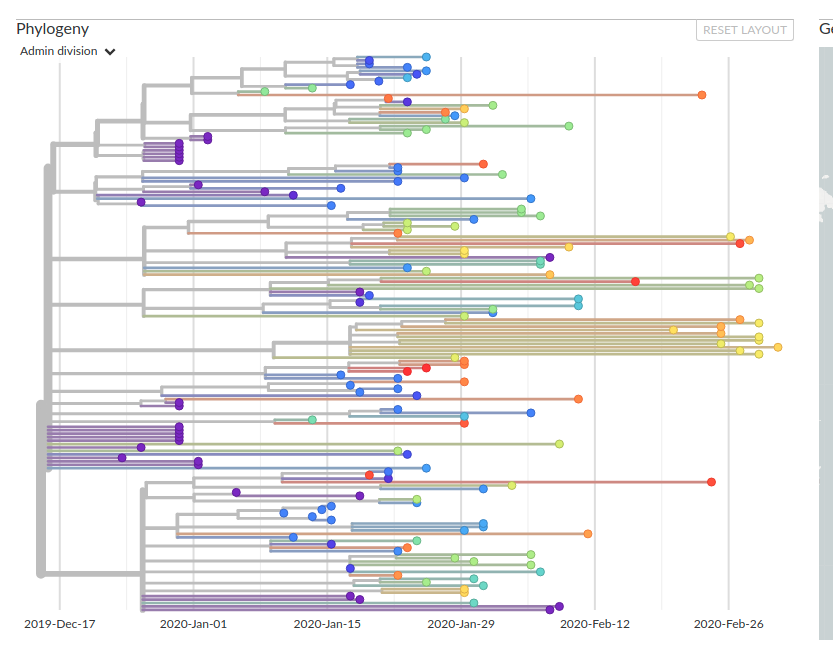
Figure 2. A screenshot of the SARS-CoV-2 time tree phylogeny from NextStrain. Colours indicate geographic location of sample. Date of sampling is shown below the tree.
The only way Tang et al. can get the results they present is by defining the ancestral state as being at some point way back in the bat coronavirus tree before the outbreak began. They then estimate the ancestral state for each mutation independently, ignoring the very informative tree of the current outbreak. This method only makes sense when using a much more closely related outgroup species, to infer the ancestral states of mutations in a freely recombinant species with unlinked mutations with independent ancestry. Whereas the most recent common ancestor of SARS-CoV-2 and the nearest bat sarbecovirus is shared many decades ago (discussed on Virological here). Additionally, such methods should incorporate the inherent uncertainty in inferring the ancestral state (e.g., est-sfs; Keightley and Jackson 2018), which Tang’s implementation does not.
Implementing this method of inferring ancestral states in a viral context, where we assume there is no recombination, means that “high frequency derived mutations” are actually just new mutations in the outbreak that have mutated back to the inferred ancestral state (in bats). This is a completely meaningless definition of “derived”. These high frequency derived mutations should instead be classed as low frequency derived mutations.
Tang et al. claim 16.3% of (7 out of 43) synonymous mutations have a derived frequency >0.5. However, given the levels of synonymous divergence, and remembering that mutations probabilities are biased, which increases the likelihood of back-mutations, this 16.3% figure is broadly in line with the expected proportion of synonymous mutations that would back-mutate to the nucleotide found in bat infecting strains. Because nonsynonymous sites are much less diverged (<4%) than synonymous sites (19%) to the most closely related bat sequence, new nonsynonymous mutations are much more likely to be away from the inferred ancestral state in bats than new synonymous mutations are. Therefore, using this flawed definition of “derived”, a much smaller proportion of nonsynonymous mutations are expected to be high frequency “derived” mutations without any action of natural selection at all.
(2) The way this data has been presented in Tang et al.’s Figure 2 will falsely suggest that purifying selection is acting even if their methodology was sensible, and there were no such selection. The height of the bars in their figure compares the raw numbers of mutations at each frequency without scaling the heights of the bars for the number of each class of mutation. Because there is a greater number of nonsynonymous polymorphisms than synonymous polymorphisms in the population, and as most mutations are expected to be at low frequency (irregardless of the action of natural selection), this presentation will always make it look like there’s proportionately more low frequency nonsynonymous mutations.
(3) When interpreting their results, Tang et al. do not consider that sequencing error could be a driver of a relative excess of singleton nonsynonymous mutations. This possibility is important because sequencing errors will be at low frequency as they are rare and cannot be transmitted, but real mutations can be at any frequency because they can be transmitted. Additionally, purifying selection can only act on real mutations, and not sequencing errors. Therefore it is very possible that sequencing error will have a higher nonsynonymous to synonymous ratio, and these mutations will be at low frequency, which will mimic the action of purifying selection suppressing the frequency of nonsynonymous mutations.
Taken together, Tang’s analysis tells us absolutely nothing about purifying selection within the viral outbreak. We have performed an additional analysis below to test for signatures of purifying selection in the SARS-CoV2 outbreak.
Additional methodological issue
The authors used the software PAML (Yang et al. 2007) to estimate selection parameters. PAML does not allow for synonymous rate variation, but they explicitly state in the paper they believe there are mutational hotspots. Recent work has shown that false positive rates of positive selection inference are unacceptably high when such synonymous rate variation occurs (Wisotsky et al. 2020). Therefore, if there truly is synonymous rate variation, to reliably identify signatures of positive selection within the phylogeny of SARS-CoV2, methods which model mutation rate variation must be used (e.g., provided by many of the models from the Hyphy package).
Summary
Given these flaws, we believe that Tang et al. should retract their paper, as the claims made in it are clearly unfounded and risk spreading dangerous misinformation at a crucial time in the outbreak.
Our Additional analysis
To test for potential purifying selection in a simple and robust manner, the number of observed synonymous and nonsynonymous mutations was compared to the null expectation by comparing the relative number of synonymous and nonsynonymous sites. The data for this analysis was taken from the CoV-GLUE resource with four samples removed from the analysis due to concern over their error rates.
The relative number of sites was estimated using the Goldman and Yang (1994) codon model. This model estimates mutation probabilities between all 61 possible coding codons using the observed frequencies of each of the 61 codons weighted by the transition to transversion ratio estimated from the data (2.9). It estimates there are 2.43 times more nonsynonymous than synonymous sites in the SARS-CoV2 genome.
This null expectation under no selection was compared to that observed from the outbreak data using a chi-squared test on the below table. This yielded a non significant P-value of 0.113. This result is not unexpected, as the current rapid growth rate of the viral population is likely to allow viruses with unfit mutations, as well as viruses with neutral mutations to be transmitted. However, we urge caution in over analysing these results, as statistical power is limited until more sequencing data accumulates.
| Nonsynonymous mutations | Synonymous mutations | |
|---|---|---|
| Null expectation | 119.7 | 49.2 |
| Observed in outbreak | 105 | 64 |
Table 1. Neutral null expectation under no selection. Adjusting for the number of sites, the point estimate for the ratio of these classes of mutations (Dn/Ds) is 0.68.
References
Foley, B., et al. “Apparent founder effect during the early years of the San Francisco HIV type 1 epidemic (1978–1979).” AIDS Research and Human Retroviruses 16.15 (2000): 1463-1469.
Goldman, N., and Ziheng Y… “A codon-based model of nucleotide substitution for protein-coding DNA sequences.” Molecular biology and evolution 11.5 (1994): 725-736.
Keightley, P. D., & Jackson, B. C. “Inferring the probability of the derived vs. the ancestral allelic state at a polymorphic site”. Genetics 209.3 (2018): 897-906.
Rai, Mohammad A., et al. “Evidence for a” Founder Effect" among HIV-infected injection drug users (IDUs) in Pakistan." BMC infectious diseases 10.1 (2010): 7.
Tsetsarkin, Konstantin A., et al. “Chikungunya virus emergence is constrained in Asia by lineage-specific adaptive landscapes.” Proceedings of the National Academy of Sciences 108.19 (2011): 7872-7877.
Wisotsky, Sadie R., et al. “Synonymous site-to-site substitution rate variation dramatically inflates false positive rates of selection analyses: ignore at your own peril.” Molecular Biology and Evolution (2020).
Yang, Ziheng. “PAML 4: phylogenetic analysis by maximum likelihood.” Molecular biology and Evolution 24.8 (2007): 1586-1591.
Acknowledgements
We would like to thank all the authors who have kindly deposited and shared genome data on GISAID. A table with genome sequence acknowledgments can be found on the CoV-GLUE website.
We thank Joseph Hughes for helpful comments.