Authors:
Carmen Lia Murall[1], Fatima Mostefai[2,3], Jean-Christophe Grenier[3], Raphaël Poujol[3], Julie Hussin[3,4], Sandrine Moreira[5], B. Jesse Shapiro[1], on behalf of the CoVSeQ consortium[6]
Affiliations:
- Department of Microbiology and Immunology, McGill University
- Département de biochimie et médecine moléculaire, Faculté de Médecine, Université de Montréal
- Research Center, Montreal Heart Institute
- Département de médecine, Faculté de Médecine, Université de Montréal
- Laboratoire de Santé Publique du Québec
- Coronavirus Sequencing in Quebec, part of the CanCOGeN network; https://covseq.ca/; CanCOGeN - GenomeCanada
Summary
- Pango lineages A, A.2.5.2 and A.27 of SARS-CoV-2 have appeared in recent outbreaks in Quebec.
- In a molecular clock analysis of global genome sequences from GISAID, we found an unexpectedly rapid accumulation of 23 nucleotide substitutions (of which 13 are missense) in A.2.5 over the first four months of the pandemic. These substitutions were inherited in descendent lineages A.2.5.1 and A.2.5.2.
- A.2.5 and descendents include the Spike missense mutations D614G and L452R, while A.27 contains L18F, N501Y, and L452R.
- Pango A.2.5 lineages likely spread north from Central America, demonstrating international spread into Canada despite border restrictions for non-essential travel.
Background
NextStrain clades 19A and 19B were the first lineages of SARS-CoV-2 discovered in humans but were largely replaced globally by clade 20 in the early months of 2020. Clade 20A and its descendants contain the spike mutation D614G, which is associated with modestly increased transmission (Volz et al. 2021). In February 2021, an apparent resurgence of clade 19B was reported. Certain lineages within clade 19B had increased in frequency, and it was hypothesized that this resurgence could be explained by convergent spike mutations that may confer fitness advantages, including S:D614G and other mutations found in SARS-CoV-2 variants of concern (VOCs) (“Resurgence of SARS-CoV-2 19B Clade Corresponds with Possible Convergent Evolution” 2021).
The CoVSeQ consortium is tasked with the sequencing of SARS-CoV-2 in the Canadian province of Quebec and has recently noticed outbreaks of clade 19B. Therefore, we present recent data from Quebec for the period Jan 2021 to Apr 1st, 2021, including ~700 new sequences deposited in GISAID (Table S1).
Results
Detection of SARS-Cov-2 Nextstrain clade 19B sequences in Quebec and globally
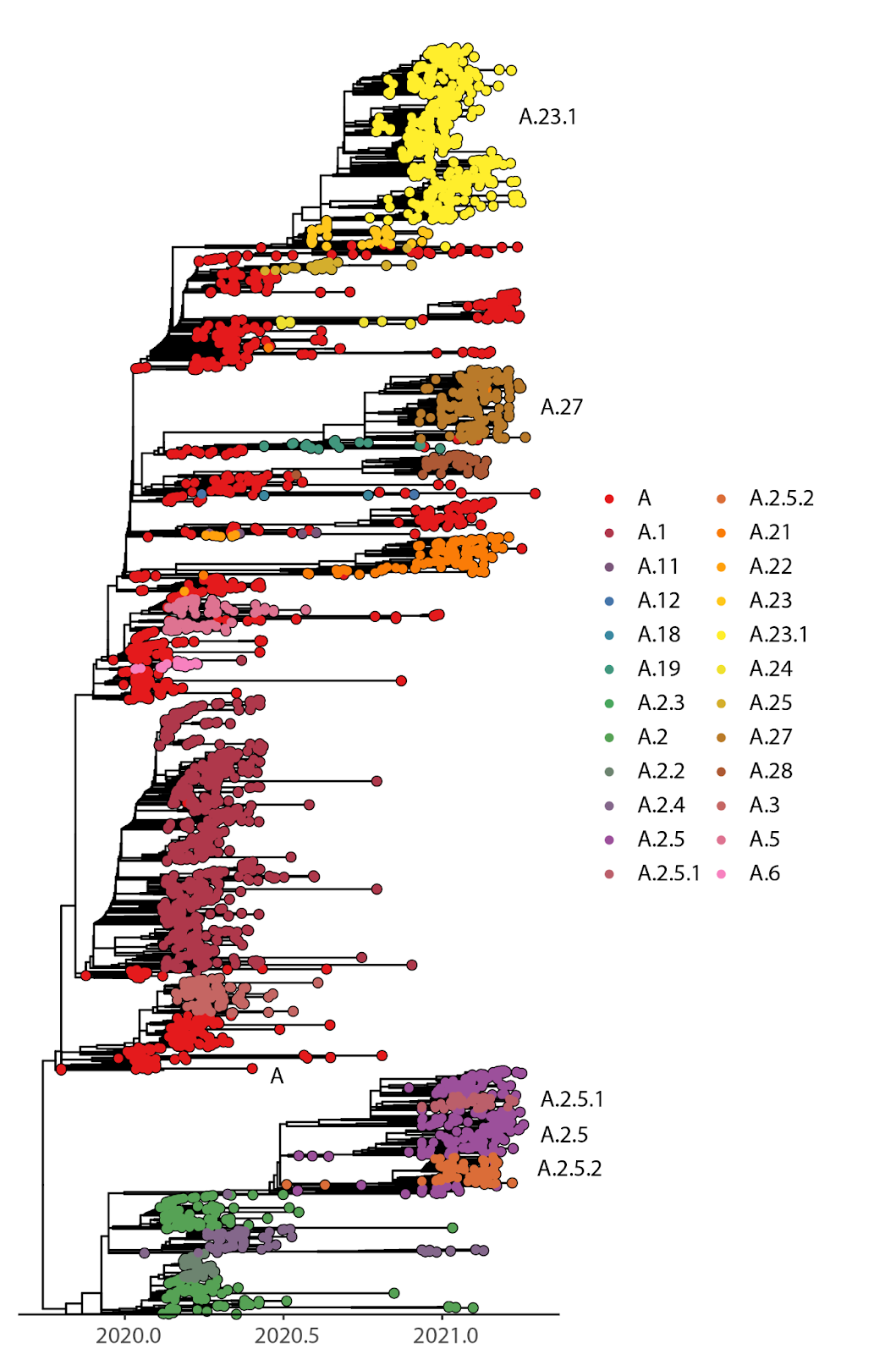
In early 2021, we found several COVID-19 cases in Quebec with sequences from Nextstrain clade 19B, specifically pango lineages A, A.2.5.2, A.21, and A.27. Globally, A.23.1, A.27, and A.2.5 (and its sublineages) are the most common clade 19B lineages observed since January 2021. Lineages A.23.1 and A.27 are distinct monophyletic clades, whereas A.2.5.1 and A.2.5.2 are sublineages nested within A.2.5 (Fig. 1).
Two outbreaks in particular (a school and long term care facility) had A.2.5.2 cases. In the long term care facility, 13 out of the 15 infected residents had been vaccinated 14 days before testing positive for SARS-CoV-2. The attack rate was greater than 30% in this facility. The school faced a large outbreak with more than 100 cases, and also had an attack rate greater than 30%. Among the 10 cases sequenced from the school, all were classified as A.2.5.2.
Fig. 1. Time-scaled phylogeny of global A lineages, including Quebec sequences. Global sequences were downsampled for easier visualization.
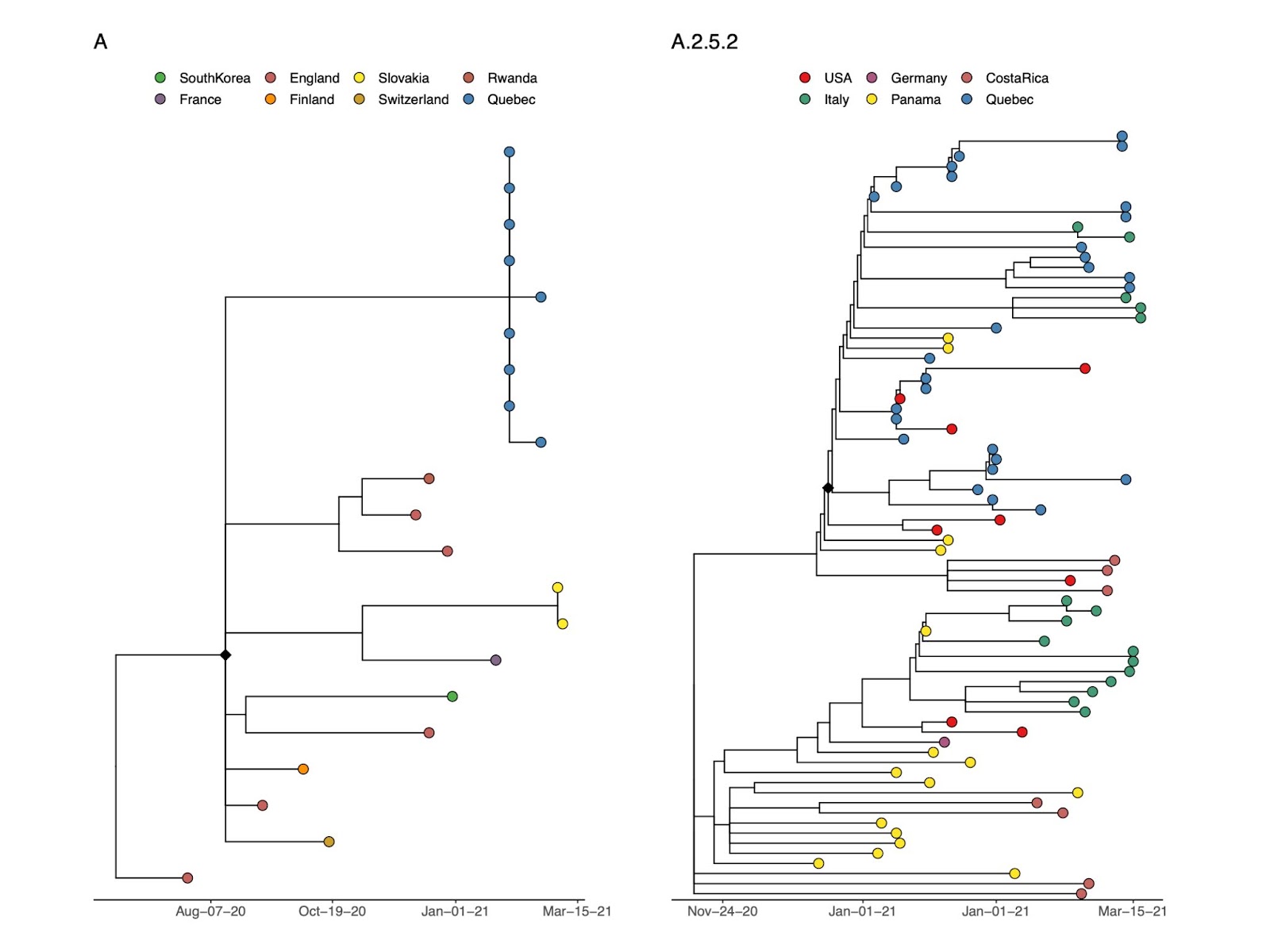
In Quebec, pango lineage A was detected in an outbreak investigation in February 2021 (sampled over two days) which was composed of near-identical sequences forming a monophyletic group arising from a polytomy, without any clear global or local origin (Fig. 2A). In contrast, pango lineage A.2.5.2 is conservatively inferred to have been introduced into Quebec at least once, likely from the USA, though the deeper branches of the global time tree are from Central America (specifically, Panama and Costa Rica), the region where this lineage was first detected (Fig. 2B).
Fig. 2. Quebec genomes of pango lineage A and A.2.5.2 in global context. Time-scaled trees are shown for pango lineage A (left) and A.2.5.2 (right). Black diamonds indicate the inferred most recent common ancestors (MRCAs) of the inferred introduction events into Quebec.
Pango lineage A.2.5 and descendants are molecular clock outliers
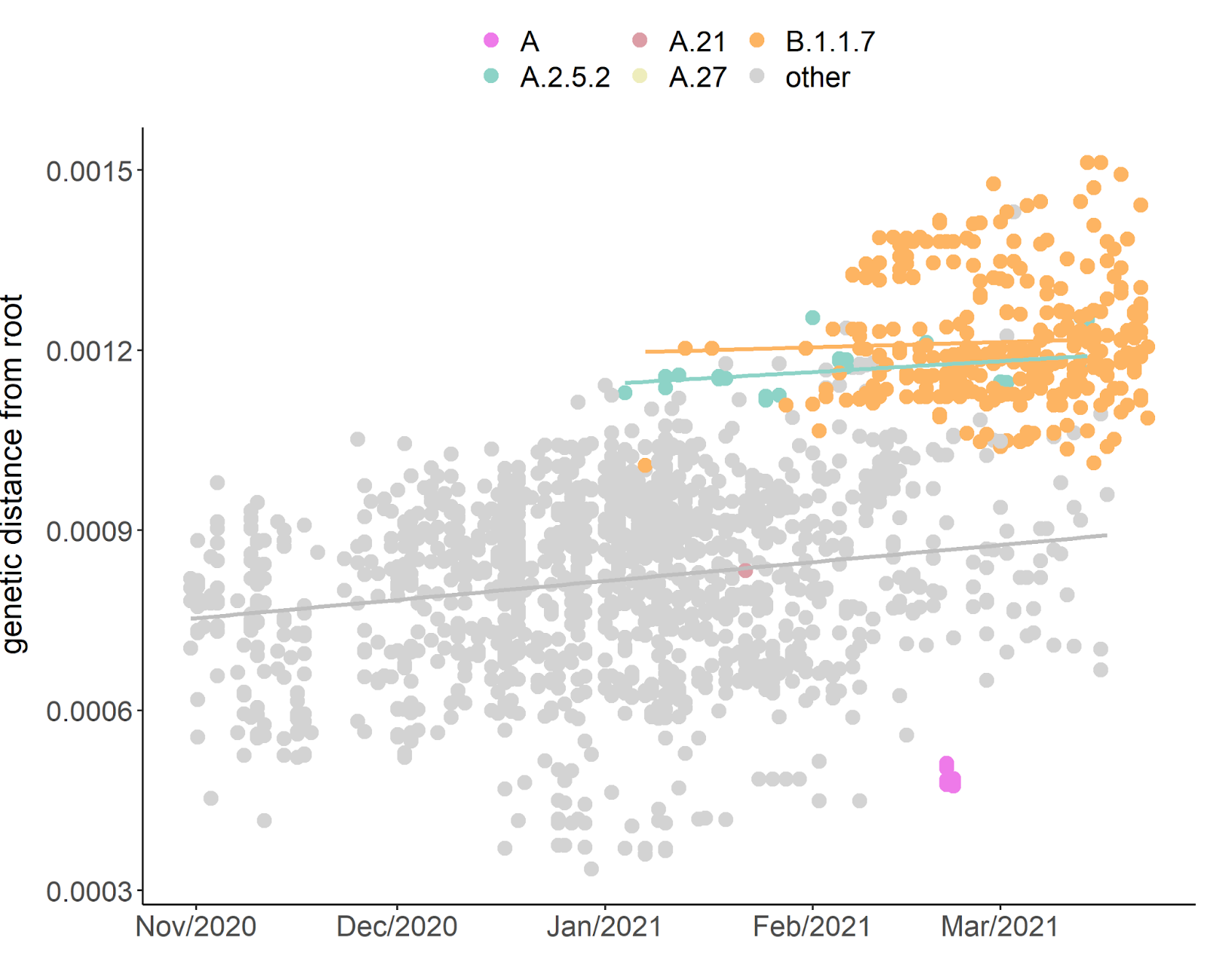
We found that A.2.5.2 (a sublineaged of A.2.5) arrived in Quebec in early January and is an outlier from the average molecular clock, with a mutational ‘jump’ of 23 substitutions, similar to that observed in B.1.1.7, a VOC in which another 23 substitutions also appeared in a short period of time (Fig. 3). B.1.1.7 was introduced into Quebec in late December 2020 and is now common across the province. While A.2.5 was introduced at a similar time, it has not increased in frequency at the same rate. Due to the higher divergence of both B.1.1.7 and A.2.5.2, the overall substitution rate in Quebec is somewhat higher with 0.0013 substitution/site/year (SE: 4.3e-05; Fig. 3) than the global average clock rate of ~0.0008 substitutions/site/year (Duchene et al. 2020; “Auspice” 2021). After its initial mutational ‘jump’ (leading to a higher intercept in the trend line), A.2.5.2 appears to evolve at an evolutionary rate similar to (or slightly slower than) other SARS-CoV-2 lineages in Quebec (Fig. 3). In contrast to A.2.5.2, pango lineage A in Quebec appears to have accumulated a relatively small number of mutations (Fig. 3).
Fig. 3. Root-to-tip divergence plot of Quebec SARS-CoV-2 sequences sampled during the second wave. Slopes represent the substitution rates of each group separately. Linear regressions: B.1.1.7: slope = 0.00011, adj R2 = -0.001, P = 0.435, A.2.5.2: slope = 0.00024, adj R2 = 0.128, P = 0.035, others: slope = 0.00038, adj R2 = 0.034, P = 4.2 x 10-13, all: slope = 0.0013, adj R2 = 0.322, P < 2.2 x 10-16.
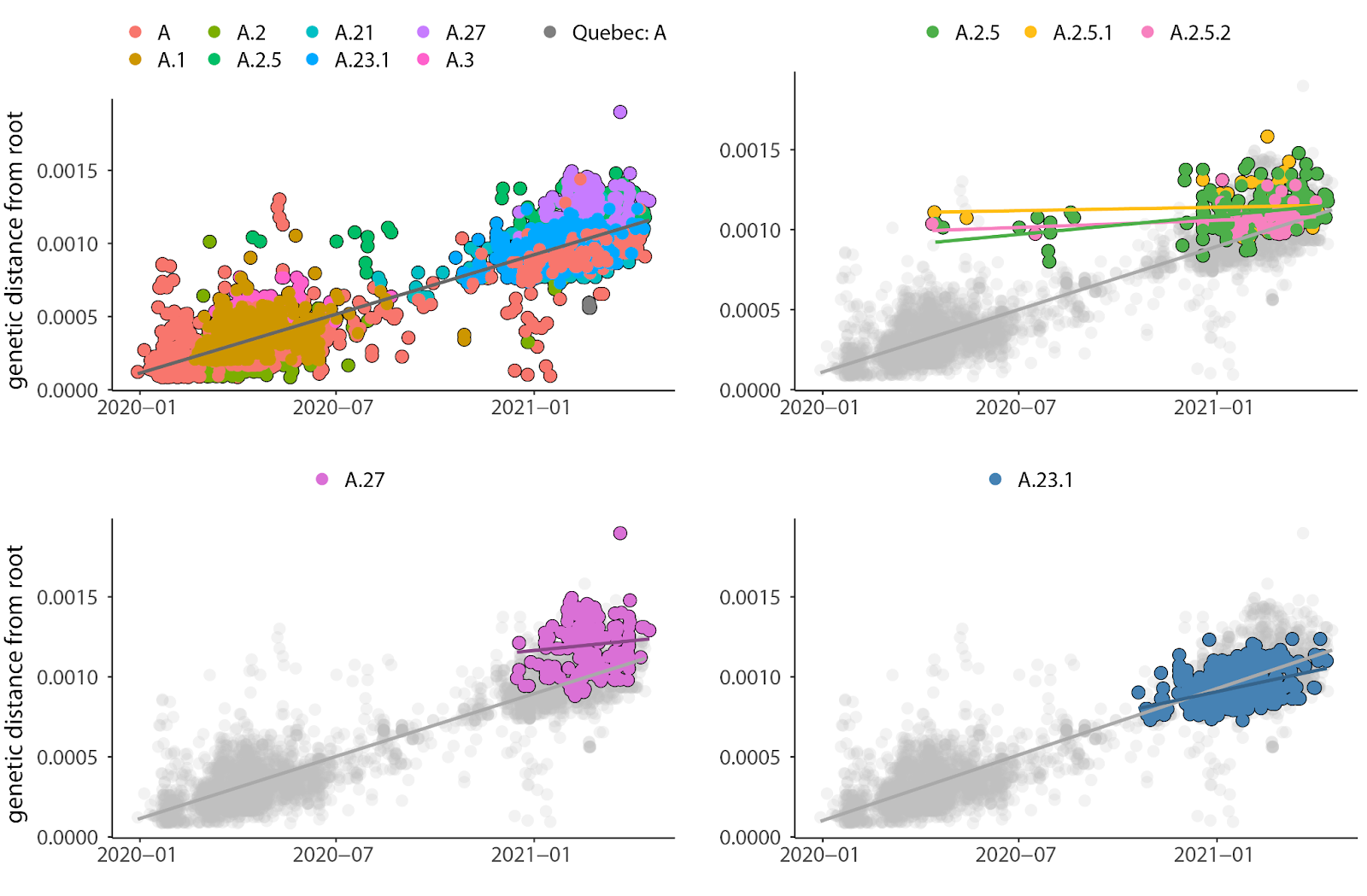
To confirm if this pattern observed in Quebec is also seen globally, we downloaded complete 19B consensus sequences with associated sampling dates from GISAID (Methods). We again found that A.2.5 (and its sublineages A.2.5.1 and A.2.5.2) experienced an excess of mutations followed by a return to the average evolutionary rate (Fig. 4). It should be noted that A.2.5 and its sublineages appeared in the first wave of the pandemic, detected mid-April 2020 in Panama (Fig. 4, top right). These sequences were flagged in GISAID as having more mutations than expected at that time but are otherwise of good quality (coverage, length, etc).
Fig. 4. Global dataset of clade 19B sequences from GISAID, including Quebec sequences. Slopes represent the substitution rates of each group considered separately. All: slope = 0.00081, se = 5.9 x 10-6 , Adj R2 = 0.835, P < 2.2 x 10-16 ; A.2.5: slope = 0.00023, se = 4.2 x 10-5, Adj R2 = 0.109, P = 0 ; A.2.5.1: slope = 0.000043, se = 1.3 x 10-4, Adj R2 = -0.024, P = 0.74 ; A.2.5.2: slope = 0.000089, se = 7.3 x 10-5, Adj R2 = 0.0088, P = 0.23 ; A.27: slope = 0.00024, se = 1.6 x 10-4, Adj R2 = 0.006, P = 0.139 ; A.23.1: slope = 0.00053, 4.9 x 10-5, Adj R2 = 0.187, P < 2.2 x 10-16.
Mutations characterising the 19B lineages
Having observed a mutational jump in pangolin lineages A.2.5 (inherited by A.2.5.1 and A.2.5.2) of similar magnitude observed in B.1.1.7, we asked which mutations appeared during this jump, and the subsequent evolution of these lineages in Quebec. As previously documented (“Resurgence of SARS-CoV-2 19B Clade Corresponds with Possible Convergent Evolution” 2021), A.2.5 acquired two spike mutations of concern, S:D614G and S:L452R, also present in the daughter lineages A.2.5.1 and A.2.5.2, but absent in the ancestral lineage A (Fig. 5). The spike mutation D614G is well-characterized, and accompanied the expansion of clade 20B in Europe in early 2020 (Volz et al. 2021). The spike mutation L452R has been observed in several other expanding lineages, including VOC B.1.617.2 lineages (Cherian et al. 2021), and is believed to cause a decreased sensitivity to neutralizing mAbs (Tada, Dcosta, et al. 2021; Tada, Zhou, et al. 2021). Furthermore, Pango lineage A.2.5 has accumulated 13 additional (compared to A) missense mutations compared to its ancestral lineage A. Lineages A.2.5.1 and A.2.5.2 inherited these 13 mutations, then A.2.5.1 acquired one additional missense mutation (NSP2:A486V), and A.2.5.2 acquired two additional missense mutations (NSP12:M502T, ORF7a:A105S). We noted a high rate of missing basecalls at genome positions 22033 and 25687 in Quebec A.2.5.2 samples (Fig. 5). It appears that the missing data at position 22033 is due to a 9-bp deletion in the primer binding region (Artic V3 amplicon 73 on the left side). The missing data at position 25687 is probably due to a mutation in the amplicon 85 primer binding region.
We note that other Quebec sequences from February 2021, assigned to pango lineage A, had acquired another independent D614G mutation in the S protein (9 sequences, Fig. 2A). This A sublineage, which also includes mutations G28878A (N:S202N/ORF9c:V49I) and G29742A (intergenic), was first been seen in Burkina Faso, Côte d’Ivoire and India in June 2020, and later in the United Kingdom (sampled in July 2020) and France (sampled in October 2020) with A.13/A.14 pango lineage assignment. All 9 Quebec sequences also include 7 additional mutations not seen in GISAID sequences from these sublineages (Fig. 5). Finally, a single sequence of A.27 was found in an outbreak investigation from late February 2021, which contains three spike mutations of interest S:L18F, S:N501Y, and S:L452R (found in VOCs B.1.1.7 and B.1.351 (Tada, Zhou, et al. 2021; Tada, Dcosta, et al. 2021)).
Fig. 5. Genomic locations of single nucleotide variants in 19B lineages of interest in GISAID and Quebec samples. Québec samples are annotated LSPQ for Laboratoire de Santé Publique du Québec. Variable nucleotide sites are listed along the x-axis in the order they appear in the genome, with the spike protein annotated with an ‘S.’ Mutations that are in at least 90% of the samples for each Pango lineage subgroup are represented as a bar, and the corresponding amino acid changes are shown on bars. All the lineages shown share the mutations C8782T (NSP4:S2839S) and T28144C (ORF8:L84S), which characterize the 19B clade.

Conclusion
Several Nextstrain clade 19B lineages appear to be resurging globally. We report on pango lineage A.2.5 and its descendants, characterized by an accumulation of a relatively large number of missense mutations, of which several in the spike protein are also observed in VOCs. Some of these mutations have been previously noted (“Resurgence of SARS-CoV-2 19B Clade Corresponds with Possible Convergent Evolution” 2021), and we expand on this work by showing an apparent ‘jump’ in evolutionary rate, similar to what was observed in B.1.1.7, followed by a return to an average mutation accumulation rate. This jump occurred sometime in early 2020, and first detected in Panama in mid-April 2020. This lineage is still currently observed in GISAID and has been implicated in some outbreaks in Quebec. It is unclear if this lineage and its descendants (A.2.5.1 and A.2.5.2) have any fitness advantage compared to other circulating lineages (e.g. B.1.1.7), but this should be assessed in ongoing surveillance. Other clade 19B lineages (specifically A.27 and A.23.1) have been declared as variants under investigation (VUI) in the UK (Public Health England 2020), and A.2.5 would also be worth monitoring closely in the future.
Acknowledgements
We thank all the authors, developers, and contributors to the GISAID database for making their SARS-CoV-2 sequences publicly available. Genome sequencing was supported by Genome Canada and Genome Quebec via CanCOGeN. BJS and CLM were supported by CoVaRR-Net. We also thank Prof. Sally Otto for constructive comments on an earlier version of this report.
Methods
We downloaded all global high quality Nextstrain clade 19B sequences (with complete sampling dates) from GISAID up to April 25th, 2021 (n = 7686), then downsampled the first wave to a maximum of 20 sequences per day chosen at random, while keeping all sequences assigned to A.2.5 (and sublineages). Additional Quebec sequences (n = 801) were added to the downsampled global dataset for a total of 3755 consensus sequences. Pango lineage assignment was run with PANGOLIN v.2.3.9 and pangoLEARN v. 2021-04-01. A multiple sequence alignment was generated with MAFFT, using Wuhan/Hu-1/2019 as the reference sequence. Maximum likelihood trees were built in IQ-Tree (with GTR model) and time trees were built using TreeTime. Clock estimates were inferred in TempEst v.1.5.3. Introduction events into Quebec were inferred on the global time tree using maximum likelihood ancestral state reconstruction of a single discrete character (QC vs. non-QC) using ace() from the ape package in R. Transition nodes were identified as QC nodes with non-QC parents and checks were performed to exclude transition nodes embedded inside later clades.
For details on sequencing and basecalling, see [Murall et al. 2021]. The complete list of GISAID IDs for all global and Quebec sequences used in this analysis can be found in Table S1. Note that some Quebec sequences (n =114) were rejected by GISAID (mostly due to a frameshift at position 28251) and thus are not included in this table. We are currently working to resolve this situation.
References
“Auspice.” Accessed May 25, 2021. auspice
Cherian, Sarah, Varsha Potdar, Santosh Jadhav, Pragya Yadav, Nivedita Gupta, Mousmi Das, Partha Rakshit, et al. 2021. “Convergent Evolution of SARS-CoV-2 Spike Mutations, L452R, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India.” bioRxiv. https://doi.org/10.1101/2021.04.22.440932.
Duchene, Sebastian, Leo Featherstone, Melina Haritopoulou-Sinanidou, Andrew Rambaut, Philippe Lemey, and Guy Baele. 2020. “Temporal Signal and the Phylodynamic Threshold of SARS-CoV-2.” Virus Evolution 6 (2): veaa061. https://doi.org/10.1093/ve/veaa061.
Murall, Carmen Lía, Eric Fournier, Jose Hector Galvez, Sarah J. Reiling, Pierre-Olivier Quirion, Sana Naderi, Anne-Marie Roy, et al. 2021. “A Small Number of Early Introductions Seeded Widespread Transmission of SARS-CoV-2 in Québec, Canada.” medRxiv, March, 2021.03.20.21253835. https://doi.org/10.1101/2021.03.20.21253835.
Public Health England. 2020. “Investigation of SARS-CoV-2 Variants of Concern: Technical Briefings.” GOV.UK. December 21, 2020. https://www.gov.uk/government/publications/investigation-of-novel-sars-cov-2-variant-variant-of-concern-20201201[.](http://paperpile.com/b/I8rDCl/oJXMO)
“Resurgence of SARS-CoV-2 19B Clade Corresponds with Possible Convergent Evolution.” 2021. February 16, 2021. https://pando.tools/t/resurgence-of-sars-cov-2-19b-clade-corresponds-with-possible-convergent-evolution/620[.](http://paperpile.com/b/I8rDCl/DGEgm)
Tada, Takuya, Belinda M. Dcosta, Marie Samanovic-Golden, Ramin S. Herati, Amber Cornelius, Mark J. Mulligan, and Nathaniel R. Landau. 2021. “Neutralization of Viruses with European, South African, and United States SARS-CoV-2 Variant Spike Proteins by Convalescent Sera and BNT162b2 mRNA Vaccine-Elicited Antibodies.” bioRxiv. https://doi.org/10.1101/2021.02.05.430003.
Tada, Takuya, Hao Zhou, Belinda M. Dcosta, Marie I. Samanovic, Mark J. Mulligan, and Nathaniel R. Landau. 2021. “The Spike Proteins of SARS-CoV-2 B.1.617 and B.1.618 Variants Identified in India Provide Partial Resistance to Vaccine-Elicited and Therapeutic Monoclonal Antibodies.” bioRxiv. https://doi.org/10.1101/2021.05.14.444076.
Volz, Erik, Verity Hill, John T. McCrone, Anna Price, David Jorgensen, Áine O’Toole, Joel Southgate, et al. 2021. “Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity.” Cell 184 (1): 64–75.e11. https://doi.org/10.1016/j.cell.2020.11.020.