RE: Proposal to redesignate B.1.631 as recombinant lineage XB
Analysis by Marina Escalera-Zamudio1,2, Hugo Gildardo Castelán Sánchez1,3, Luis José Delaye Arredondo1,4, Bernardo Gutiérrez1,2,5
1Department of Zoology, University of Oxford, Oxford, UK
2Consorcio Mexicano de Vigilancia Genómica (CoViGen-Mex)*
3CONACYT-UAEM-Centro de Investigación en Dinámica Celular
4Departamento de Ingeniería Genética, Unidad Irapuato, CINVESTAV, Mexico
5Colegio de Ciencias Biológicas y Ambientales, Universidad San Francisco de Quito USFQ, Quito, Ecuador
Correspondence to: [email protected]
*For details on data generated in Mexico, see the Methods section.
Frequent recombination observed among coronaviruses circulating in the human population suggests that this could be a common evolutionary mechanism that could lead to the emergence new SARS-CoV-2 variants1. Recombinant SARS-CoV-2 genomes involving lineage B.1.1.7 have been detected in the UK2, whilst several recombinant SARS-CoV-2 variants have been detected to currently circulate at low levels3. Following the preliminary results and proposal previously raised4, we performed detailed phylogenetic analysis to detect recombinant sequences/lineages and any putative breakpoints within the virus genome bases on topology congruency and SNP distribution (see Methods). The same general phylogenetic pattern was observed for the whole genome (WG) and Orf1ab trees, whilst the S and 3’-UP also showed comparable phylogenetic patterns between them. However, phylogenetic incongruencies were observed between the WG and Orf1ab trees, compared to the S and 3’-UP trees (Figure 1a). By comparing the AICc score of the best fitting model allowing for different topologies between segments, using GARD (see Methods), one breakpoint was identified which reflects a true topological incongruence (alignment site 22490, 562.098 Δ c-AIC vs the null model; Figure 1b). This site corresponds to codon 22775-22778 [GAT] in the Spike Orf; it corresponds to amino acid residue 497D, located in the trimer interface of S close to a putative receptor binding site 5 (in reference to the Wuhan-Hu-1 genome numbering; Figure 1b). Recombinant sequences identified under 3Seq and RDP4 (628_USA_CO-CDC-MMB09514673_2021_EPI_ISL_3428959_20 and 628_USA_MN-MDH-9090_2021_EPI_ISL_3260223_2021-07-2) precede the B.1.631 recombinant cluster in the Spike and 3’-UP trees (marked with an *, most basal sequence for B.1.631: 631_Mexico_CHH_IBT_IMSS_1405_2021_EPI_ISL_3355598_2021-05-27; Figures 1a, 1c). SNPs shared between B.1.628 and B.1.631 are shown in Figure 1c. Thus, we support the proposal to redesignate B.1.631 as recombinant lineage, as we find evidence for recombination for a large cluster of sequences comprising the majority of the B.1.631 lineage.
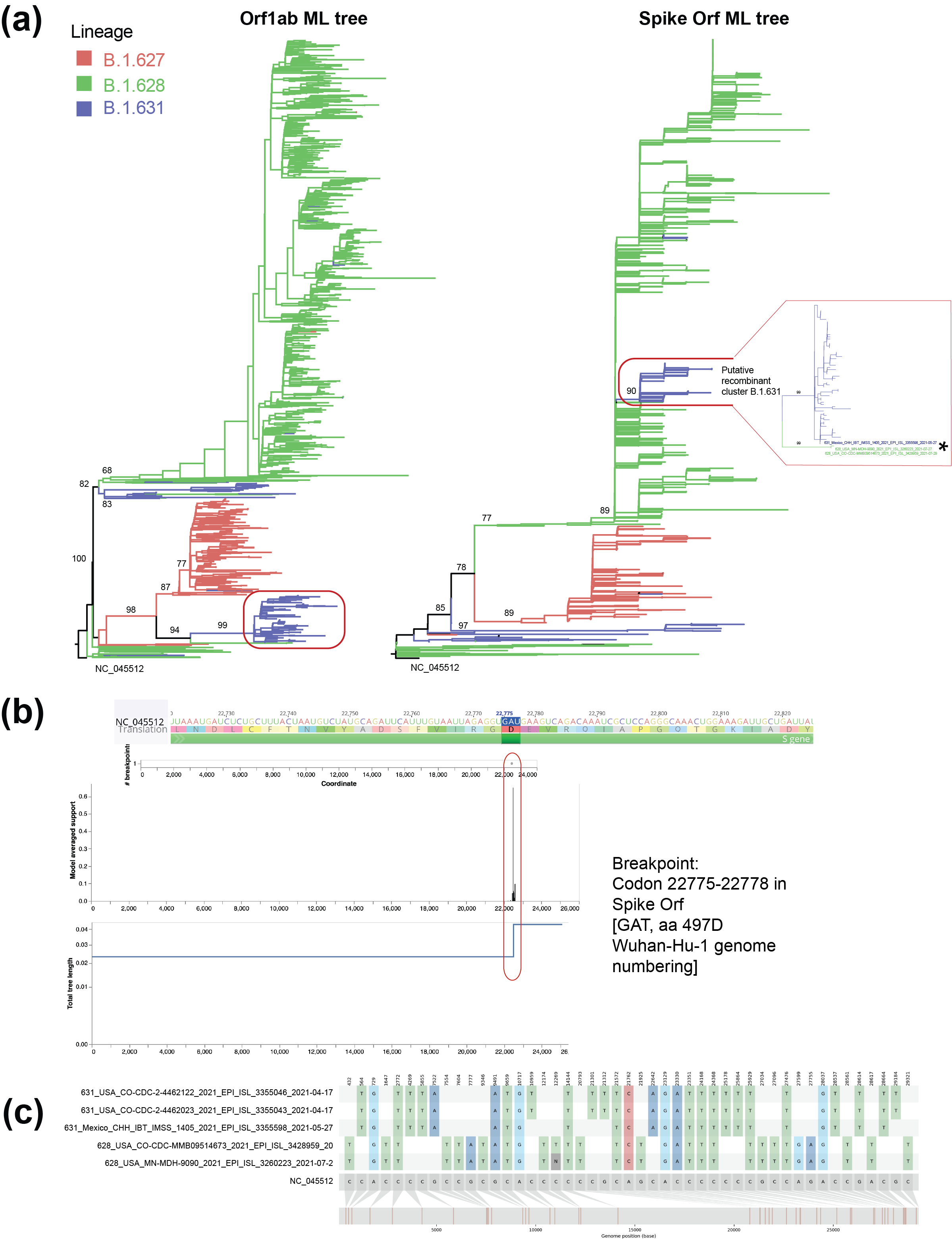
Figure 1. (a) Maximum likelihood phylogenetic trees of two SARS-CoV-2 genome partitions for the B.1.631, B.1.627 and B.1.628 lineages. (b) Recombination analysis performed on the whole genome alignment (for the aforementioned lineages) using GARD. (c) Single nucleotide polymorphisms (SNPs) shared between lineages B.1.628 and B.1.631.
Methods
Genomic dataset
Complete genome datasets available in GSAID6 for the B.1.627 (239 sequences in Cov-Lineages7, representative sequence USA/MN-UMGC-3398/2021; 2021-04-24), B.1.628 (1095 sequences in Cov-Lineages7, representative sequence USA/LA-BIE-LSUH000937/2021; 2021-06-19) and B.1.631 (154 sequences in Cov-Lineages7, representative sequence USA/NY-PRL-2021_0419_02G09/2021) virus lineages were downloaded as of 2021-08-17 (1487 sequences in total). From these, 415 of the sequences derive from Mexican cases (47 for B.1.627, 340 for B.1.628 and 27 for B.1.631), of which 409 were generated as part of the baseline/sentinel surveillance national programme by InDRE (Institute of Epidemiological Diagnosis and Reference, Ministry of Health, Mexico) and the Consorcio Mexicano de Vigilancia Genómica (CoViGen-Mex)8. The remaining 6 sequences derive from non-sentinel-surveillance sampling (hospital/laboratory).
Initial phylogenetic analysis
For each dataset, sequences were mapped to the complete genome of SARS-CoV-2 isolate Wuhan-Hu-1 (NC_045512) using Geneious (https://www.geneious.com/) to generate individual alignments that were visually inspected for accuracy. Sequences were removed from the datasets if (i) they were >1000nt shorter than full genome length, (ii) they were 100% similar to any other sequence, or (iii) if >10% of site were ambiguities (including N or X). Each alignment was partitioned into three sub-alignments according to different genomic regions: Orf1ab, S and and the 3’ upstream region’ (3’-UP; encompassing all coding sequences after Spike, including Orf3 up to before the 3’ UTR). The partitioned and whole genome alignments were used to estimate four initial large-scale Maximum Likelihood phylogenies (LS WG, Orf1ab, S and 3’-UP) using IQ-TREE (http://www.iqtree.org/) under a general time reversible nucleotide substitution model with gamma-distributed among-site rate variation (GTR+G); branch support was assessed using 100 bootstrap replicates. All trees were rooted using the basal NC_045512 node.
Detection of recombinant sequences/clusters
The initial large-scale Maximum Likelihood phylogenies were used to detect general phylogenetic pattern incongruencies by comparing the tree topologiess obtained for the different genomic regions. Large-scale trees were used for further subsampling the whole genome alignment in a phylogenetically-informed manner, in order to normalise the number of sequences representing each virus lineage and to reduce alignment size by removing non-informative sequences. The subsampled whole genome alignment (197 sequences, 25095 sites) was analysed using GARD (Genetic Algorithm for Recombination Detection) in HYPHY9. The subsampled alignment was also analysed using 3Seq and RDP410,11.
References
1 Li, X. et al. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci Adv 6, doi:10.1126/sciadv.abb9153 (2020).
2 Jackson, B. et al. Recombinant SARS-CoV-2 genomes involving lineage B.1.1.7 in the UK, https://pando.tools/t/recombinant-sars-cov-2-genomes-involving-lineage-b-1-1-7-in-the-uk/658/1 (2020).
3 VanInsberghe, D., Neish, A. S., Lowen, A. C. & Koelle, K. Recombinant SARS-CoV-2 genomes are currently circulating at low levels. bioRxiv, doi:10.1101/2020.08.05.238386 (2021).
4 Jackson, B. Proposal to redesignate B.1.631 as recombinant lineage XB #189, https://github.com/cov-lineages/pango-designation/issues/189 (2021).
5 NCBI RefSeq SARS-CoV-2 genome graphical display, https://www.ncbi.nlm.nih.gov/projects/sviewer/?id=NC_045512 (NCBI).
6 GISAID. Global Initiative on Sharing Avian Influenza Data, https://www.gisaid.org/ (2021).
7 O’Toole, A. et al. Tracking the international spread of SARS-CoV-2 lineages B.1.1.7 and B.1.351/501Y-V2, https://pando.tools/t/tracking-the-international-spread-of-sars-cov-2-lineages-b-1-1-7-and-b-1-351-501y-v2/592 (2021).
8 Genómica, C. M. d. V. CoViGen-Mex, http://mexcov2.ibt.unam.mx:8080/COVID-TRACKER/
9 Kosakovsky Pond, S. L., Posada, D., Gravenor, M. B., Woelk, C. H. & Frost, S. D. GARD: a genetic algorithm for recombination detection. Bioinformatics 22, 3096-3098, doi:10.1093/bioinformatics/btl474 (2006).
10 Martin, D. P., Murrell, B., Golden, M., Khoosal, A. & Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol 1, vev003, doi:10.1093/ve/vev003 (2015).
11 Lam, H. M., Ratmann, O. & Boni, M. F. Improved Algorithmic Complexity for the 3SEQ Recombination Detection Algorithm. Mol Biol Evol 35, 247-251, doi:10.1093/molbev/msx263 (2018).