Due to length constraints on virological.org posts, we are splitting this report into two parts that should be read as a single report. This is Part 2, containing Findings sections 6 through 9, Concluding remarks, Acknowledgements, and References. For Background, Data, Methods, and Findings sections 1 through 5. See Part 1
Michael Worobey, University of Arizona, Tucson, AZ 85705, USA
Karthik Gangavarapu, Department of Immunology and Microbiology, The Scripps Research Institute, La Jolla, CA, USA
Jonathan E. Pekar, Department of Medicine, University of California San Diego, La Jolla, CA, 92093, USA
Jeffrey B. Joy, Department of Medicine, University of British Columbia, Vancouver, BC, Canada
Louise Moncla, University of Pennsylvania, PA 19104, USA
Moritz U. G. Kraemer, Department of Biology & Pandemic Sciences Institute, University of Oxford, Oxford, UK
Gytis Dudas, Institute of Biotechnology, Life Sciences Center, Vilnius University, Vilnius, Lithuania
Daniel Goldhill, Department of Pathobiology and Population Sciences, Royal Veterinary College, London, UK.
Christopher Ruis, VPD Heart & Lung Research Institute, Department of Medicine, University of Cambridge, Cambridge, UK
Lorena Malpica Serrano, University of Arizona, Tucson, AZ 85705, USA
Xiang Ji, Department of Mathematics, Tulane University, New Orleans, LA 70118, USA
Kristian G. Andersen, Department of Immunology and Microbiology, The Scripps Research Institute, La Jolla, CA, USA
Joel O. Wertheim, Department of Medicine, University of California San Diego, La Jolla, CA, 92093, USA
Philippe Lemey, Department of Microbiology, Immunology and Transplantation, Rega Institute, KU Leuven, Leuven, Belgium
Marc A Suchard, Department of Biostatistics, University of California, Los Angeles, Los Angeles, CA 90095, USA
Angela L. Rasmussen, Vaccine and Infectious Disease Organization, University of Saskatchewan, Saskatoon, SK, Canada S7N 5E3
Meera Chand, UK Health Security Agency, London UK
Natalie Groves, UK Health Security Agency, London UK
Oliver G. Pybus, (1) Department of Pathobiology and Population Sciences, Royal Veterinary College, London, UK (2) Department of Biology & Pandemic Sciences Institute, University of Oxford, Oxford, UK
Thomas P. Peacock, The Pirbright Institute, Woking, UK, GU24 0NF; Department of Infectious Disease, Imperial College London, UK, W2 1PG
Andrew Rambaut, Institute of Ecology and Evolution, University of Edinburgh, Edinburgh, UK
Martha I. Nelson, National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20892
6. The original cattle H5N1 virus’s HA was not adapted to a human-like receptor.
- In the hemagglutinin (HA), the protein that must bind to the host’s cell-surface sialic acid residues for the virus to gain entry to the host cell, the closest-related sequences to the cattle H5N1 HAs come from wild birds (a Canada goose and a peregrine falcon.) There are no amino acid differences between the HAs of these wild bird sequences and the earliest sequences from cattle.
- This suggests that the first cattle sequences possessed no (pre)adaptation to mammalian cell-surface receptors. Non-synonymous changes in HA have been acquired by small clades of sequences from cattle and cats nested within the cattle clade (n<=11); however, the significance of these mutations is not well understood. Although mutations have been seen in the polymerase of viruses from cattle, similar mutations have been seen in avian viruses infecting humans and other mammals without leading to sustained transmission. Without changes in HA affecting receptor binding, the risk of the virus becoming transmissible between humans is low. The lack of HA changes may also suggest (at least with the current tissue tropism) that there is not strong selective pressure to change receptor binding, suggesting ‘avian-like’ α-2,3-linked sialic acids are abundant in the main sites of replication in these animals. However, this is also true for dogs and pigs, which in the short term do not strongly select for such changes, yet in the longer term, avian-origin H1N1 and H3N2 viruses in these species gradually adapt to ‘human-like’ α-2,6-linked sialic acids(37)(38)
7. H5N1 is transmitting from cattle back into wild birds, poultry, cats, and other species.
- Several sequences from wild birds (e.g., blackbird, grackle), poultry, domestic cats, and other wildlife (e.g., raccoon) are nested within the main cattle clade of sequences across each genome segment (Figures 2 & 3; see Figure 7 for concatenated genome tree). This tree topology suggests that the virus may be spilling back from cows into other host species, which is consistent with prior reports of virus transmission between cattle and poultry at individual farms(39, 40) [https://www.doi.org/10.1126/science.zoo2sbi]and from cows into cats (15) .
- While most “spillbacks” from cattle were observed in Texas, one cattle-to-poultry transmission was observed in Michigan (clustering with Michigan cattle viruses), and another spillback to raccoons occurred in New Mexico (clustering with New Mexico cattle viruses).
- Whether any onward cat-to-cat or poultry-to-poultry transmission occurred following these spillbacks is difficult to resolve at this time. But these data suggest that H5N1 transmission in cattle is extensive enough to initiate outbreaks in other host species.
- Further evidence of spillback from cattle to other species comes from amino acid changes that are fixed in cattle that are also seen in these birds and other mammals, including two putative mammalian adaptations PB2 M631L and PA K497R. Assuming a single spillover from wild birds into cows, the viruses have likely spread locally from cows back into wild birds, which associate with cattle on farms.
- There have been at least two independent spillbacks to domestic (presumably barn) cats in Texas, most likely due to the consumption of raw milk from infected dairy cattle. H5N1 is known to be highly pathogenic in cats, and early on, dead cats served as sentinels on dairy farms when the symptoms in cattle were less severe and less HPAI-specific (41).
8. A virus closely related to, but distinct from, those sampled from cattle was sampled from an individual who was reportedly a dairy farm worker.
- We estimated the tMRCA of the cattle clade and the human virus as 22 November 2023 (95% HPD: [26 September 2023, 19 January 2024]). The human sequence (A/Texas/37/2024) does not share multiple substitutions present in all the cattle sequences (Figure 7 clades A-E).
- Moreover, the A/Texas/37/2024 strain contains four unique amino acid changes in PB2, PB1, PA, and NS1 that are not found in the cattle sequences or in the basal avian/wildlife sequences. The human virus is separated by a long branch length that likely represents several weeks of unsampled diversity.
- Therefore, we can’t currently resolve whether (a) the human sequence is a descendent of an unsampled cattle lineage that branched off early from the (single-origin) cattle H5N1 clade shortly after its establishment, or (b) emerged after a separate jump from the avian reservoir into cattle, and then from cattle to that individual on the farm where the person worked, or (c) is a direct spillover from an avian source. Scenario (c) seems highly unlikely. Scenarios (a) and (b) would imply unsampled diversity along that lineage in cattle perhaps due to lower fitness of that lineage in cattle(42), or overdispersion of the cattle lineage due to it entering a more highly connected network of hosts.
- There are currently no cattle sequences from the farm where this case occurred (our understanding is that none have been collected there, or that the specific farm might not be known). Nor do we have data on cattle movements which could help distinguish between these possibilities.
- We also do not know which of the four mutations seen in the human virus emerged in that individual during replication, versus upstream, in cattle or birds that were never sampled. E627K in PB2 is a mammalian adaptation that is often selected for during host replication cycles when H5N1 spills over into mammals (e.g., seal, fox, raccoon, skunk), and it is possible E627K emerged in the human in a similar fashion, raw sequence files from the human case could help resolve this.
- The human sequence or its most recent common ancestor with the cattle is basal to the cattle viruses in HA, NP, PB2, and PA, but it has derived and/or private nucleotide substitutions in other segments, thereby making it challenging to resolve a clear transmission chain or exclude the possibility of reassortment. For instance, there is an A->G substitution in NA that is shared between the human H5N1 virus and 3 cattle virus sequences (SRR28752670, SRR28752678, and SRR28752626).
9. Mutational signatures support sustained transmission within cattle.
- We aimed to determine whether we could identify host species transmission routes in the cattle outbreak using mutational signatures, which have previously been shown to be influenced by transmission route and host species(43–45). We calculated mutational spectra for three groups of host species within the complete H5 goose-Guangdong lineage: wild birds, chickens and mammals.
- Comparison of the mutational signatures between these host species groups showed T-to-C mutations to be significantly elevated in mammals compared with wild birds (Figure 8).
- We then calculated the mutational spectrum of the cattle outbreak and found that this shows high levels of T-to-C mutations, consistent with transmission amongst mammals. We then compared the likelihoods of generating the set of mutations observed in the cattle outbreak under the mutational spectra of mammals, wild birds and chickens. We found that the mutations were much more likely to have been generated under the mammal spectrum compared to the wild bird spectrum (probability of mammal 0.997) and compared to the chicken spectrum (probability of mammal 0.94) (Figure 8b).
- Examining only mutations on the internal phylogenetic branches of the cattle outbreak again showed support for mammals over wild birds (probability of mammal 0.89) but the comparison between mammal and chicken spectra was inconclusive (probability of mammal 0.49) (Figure 8c). This is potentially due to the small number of sampled mutations on the internal phylogenetic branches (n = 146); we often observe intermediate probabilities for the known mammal, wild bird and chicken spectra at this number of mutations (Figure 8c).
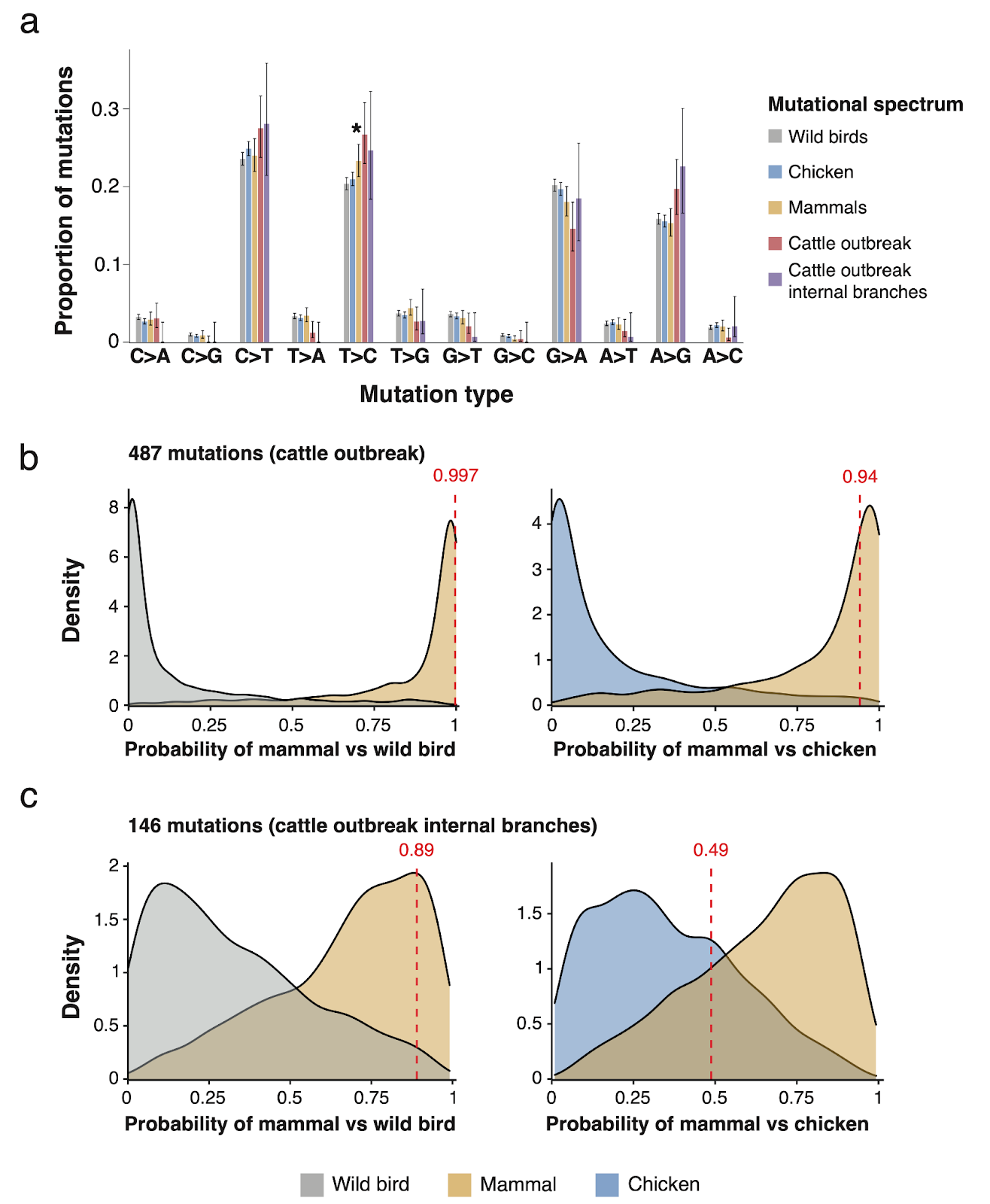
Figure 8. Examination of mutational signatures. a, comparison of the proportion of total mutations that are of each mutation type (C>A for example refers to C mutating to A) across different mutational spectra. * shows that T>C mutations occur significantly more often in the mammal spectrum compared to the wild bird spectrum. b and c the probability of generating the observed mutations in the full cattle outbreak clade (b) and its internal phylogenetic branches (c) under the mammal mutational spectrum compared to either the wild bird or chicken spectrum are shown in red. The distributions show the same probability for 1000 random subsamples of the respective mutational spectra to the same number of mutations.
Concluding remarks
We believe that it is essential to define a comprehensive genomic and serological surveillance strategy for this outbreak and future ones. Such a strategy would (i) incorporate samples from mammals, birds and people, including for example milk and wastewater; (ii) aim to define the source of the outbreak; and (iii) monitor the spread, trajectory, and evolution of the virus, and its pandemic potential.
We urge the transparent, timely, and public sharing of data from various sources, including sequencing data, number of tests being performed, dates when samples were taken, locations of sampling and, when possible, transportation history of cattle. Such integrated data would help the scientific and animal/public health community best understand the dynamics of these viruses among hosts, and through time and space, and may thereby lead to timely interventions to mitigate its spread, not just among cattle and cattle operations but also more broadly.
Acknowledgements
We thank Andrew Bowman, Jim Lowe, and Richard Webby for sharing H5N1 sequences from cattle from Ohio; USDA-APHIS for sharing sequence read data on SRA; Florence Débarre for procuring sampling dates and locations for many of these viruses; Trevor Bedford for assistance creating the tanglegram in Figure 1; and GISAID contributors for sharing H5N1 data. An acknowledgements table of GISAID contributors is at avian-influenza/acknowledgements/gisaid_acknowledge_table_assemby_reference_sequences.xls at master · andersen-lab/avian-influenza · GitHub. The views expressed are the authors’ and do not necessarily represent the views of the National Institutes of Health or the United States Government.
References
-
G. Tomás, A. Marandino, Y. Panzera, S. Rodríguez, G. L. Wallau, F. Z. Dezordi, R. Pérez, L. Bassetti, R. Negro, J. Williman, V. Uriarte, F. Grazioli, C. Leizagoyen, S. Riverón, J. Coronel, S. Bello, E. Páez, M. Lima, V. Méndez, R. Pérez, Highly pathogenic avian influenza H5N1 virus infections in pinnipeds and seabirds in Uruguay: implications for bird-mammal transmission in South America. Virus Evol, doi: 10.1093/ve/veae031.
-
E. R. Burrough, D. R. Magstadt, B. Petersen, S. J. Timmermans, P. C. Gauger, J. Zhang, C. Siepker, M. Mainenti, G. Li, A. C. Thompson, P. J. Gorden, P. J. Plummer, R. Main, Early Release - Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4b Virus Infection in Domestic Dairy Cattle and Cats, United States, 2024 - Volume 30, Number 7—July 2024 - Emerging Infectious Diseases journal - CDC. doi: 10.3201/eid3007.240508.
-
Highly Pathogenic Avian Influenza Scientific Symposium (2024; https://www.youtube.com/watch?v=aTizSNagjFI[).](http://paperpile.com/b/DYND7R/vQ2J)
-
avian-influenza/metadata/PRJNA1102327_metadata.csv at master · andersen-lab/avian-influenza, GitHub. https://github.com/andersen-lab/avian-influenza/blob/master/metadata/PRJNA1102327_metadata.csv[.](http://paperpile.com/b/DYND7R/OI9S)
-
G. Dudas, Baltic: Baltic - Backronymed Adaptable Lightweight Tree Import Code for Molecular Phylogeny Manipulation, Analysis and Visualisation. Development Is Back on the Evogytis/baltic Branch (i.e. Here) (Github; https://github.com/evogytis/baltic[).](http://paperpile.com/b/DYND7R/FvhY)
-
H. Branswell, Genetic analysis reveals H5N1 flu virus outbreak in cows likely started earlier than thought, STAT (2024). https://www.statnews.com/2024/04/23/h5n1-bird-flu-genetic-analysis/[.](http://paperpile.com/b/DYND7R/jC9j)
-
Center for Food Safety, A. Nutrition, Updates on Highly Pathogenic Avian Influenza (HPAI), U.S. Food and Drug Administration (2024). https://www.fda.gov/food/alerts-advisories-safety-information/updates-highly-pathogenic-avian-influenza-hpai[.](http://paperpile.com/b/DYND7R/Lyc4)
-
E. Fodor, E. Staller, L. Carrique, O. Swann, H. Fan, J. Keown, C. Sheppard, W. Barclay, J. Grimes, Structures of H5N1 influenza polymerase with ANP32B reveal mechanisms of genome replication and host adaptation. doi: 10.21203/rs.3.rs-3716220/v1 (2024).
-
U.S. government in hot seat for response to growing cow flu outbreak, American Association for the Advancement of Science (AAAS) (2024); https://doi.org/10.1126/science.zoo2sbi.
-
ABC News, This Texas veterinarian helped crack the mystery of bird flu in cows, ABC News (2024). https://abcnews.go.com/Health/wireStory/texas-veterinarian-helped-crack-mystery-bird-flu-cows-109836066[.](http://paperpile.com/b/DYND7R/LBLu)
-
Website. https://twitter.com/MichaelWorobey/status/1783992353595990498[.](http://paperpile.com/b/DYND7R/KqTh)