Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations
Report written by: Andrew Rambaut1, Nick Loman2, Oliver Pybus3, Wendy Barclay4, Jeff Barrett5, Alesandro Carabelli6, Tom Connor7, Tom Peacock4, David L Robertson8, Erik Volz4, on behalf of COVID-19 Genomics Consortium UK (CoG-UK)9.
- University of Edinburgh
- University of Birmingham
- University of Oxford
- Imperial College London
- Wellcome Trust Sanger Institute
- University of Cambridge
- Cardiff University
- MRC-University of Glasgow Centre for Virus Research
- https://www.cogconsortium.uk
![]()
This work is licensed under a Creative Commons Attribution-ShareAlike 4.0 International License.
Summary
Recently a distinct phylogenetic cluster (named lineage B.1.1.7) was detected within the COG-UK surveillance dataset. This cluster has been growing rapidly over the past 4 weeks and since been observed in other UK locations, indicating further spread.
Several aspects of this cluster are noteworthy for epidemiological and biological reasons and we report preliminary findings below. In summary:
The B.1.1.7 lineage accounts for an increasing proportion of cases in parts of England. The number of B.1.1.7 cases, and the number of regions reporting B.1.1.7 infections, are growing.
B.1.1.7 has an unusually large number of genetic changes, particularly in the spike protein.
Three of these mutations have potential biological effects that have been described previously to varying extents:
- Mutation N501Y is one of six key contact residues within the receptor-binding domain (RBD) and has been identified as increasing binding affinity to human and murine ACE2.
- The spike deletion 69-70del has been described in the context of evasion to the human immune response but has also occurred a number of times in association with other RBD changes.
- Mutation P681H is immediately adjacent to the furin cleavage site, a known location of biological significance.
The rapid growth of this lineage indicates the need for enhanced genomic and epidemiological surveillance worldwide and laboratory investigations of antigenicity and infectivity.
Background
The two earliest sampled genomes that belong to the B.1.1.7 lineage were collected on 20-Sept-2020 in Kent and another on 21-Sept-2020 from Greater London. B.1.1.7 infections have continued to be detected in the UK through early December 2020. Genomes belonging to lineage B.1.1.7 form a monophyletic clade that is well supported by a large number of lineage-defining mutations (Figure 1). As of 15th December, there are 1623 genomes in the B.1.1.7 lineage. Of these 519 were sampled in Greater London, 555 in Kent, 545 in other regions of the UK including both Scotland and Wales, and 4 in other countries.
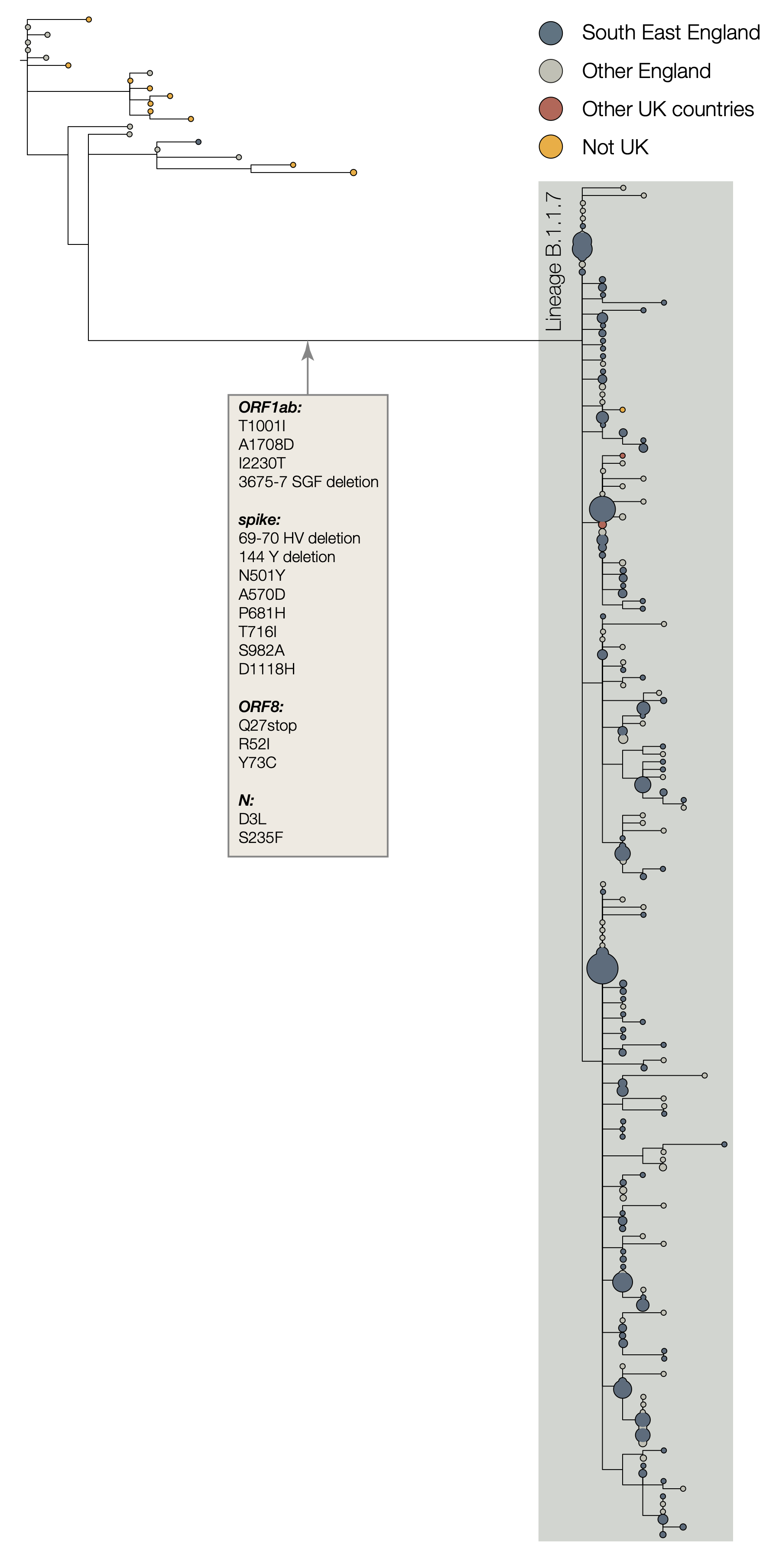
Figure 1 | Phylogenetic tree of the B.1.1.7 lineage and its nearest outgroup sequences, for samples collected up until 30-Nov-2020. Tips from the same location have been collapsed into circles whose area is proportional to the number of genomes represented. Three large subclades are evident within the B.1.1.7 lineage, each defined by one nucleotide change. One of these clades is defined by a further stop codon in ORF8.
Lineage-defining mutations & rate of evolution
The B.1.1.7 lineage carries a larger than usual number of virus genetic changes. The accrual of 14 lineage-specific amino acid replacements prior to its detection is, to date, unprecedented in the global virus genomic data for the COVID-19 pandemic. Most branches in the global phylogenetic tree of SARS-CoV-2 show no more than a few mutations and mutations accumulate at a relatively consistent rate over time. Estimates suggest that circulating SARS-CoV-2 lineages accumulate nucleotide mutations at a rate of about 1-2 mutations per month (Duchene et al. 2020).
A preliminary analysis of these observations is provided in Figure 2, which shows a regression of root-to-tip genetic distances against genome sampling date, for lineage B.1.1.7 and for a selection of related outgroup genomes. The rate of molecular evolution within lineage B.1.1.7 is similar to that of other related lineages. However, lineage B.1.1.7 is more divergent from the phylogenetic root of the pandemic, indicating a higher rate of molecular evolution on the phylogenetic branch immediately ancestral to B.1.1.7. Further, inferred nucleotide changes on this branch are predominantly amino acid-altering (14 non-synonymous mutations and 3 deletions). There are 6 synonymous mutations on the branch. This is suggestive of a process involving adaptive molecular evolution, although a role for increased fixation rates through relaxed selective constraint cannot be currently ruled out.
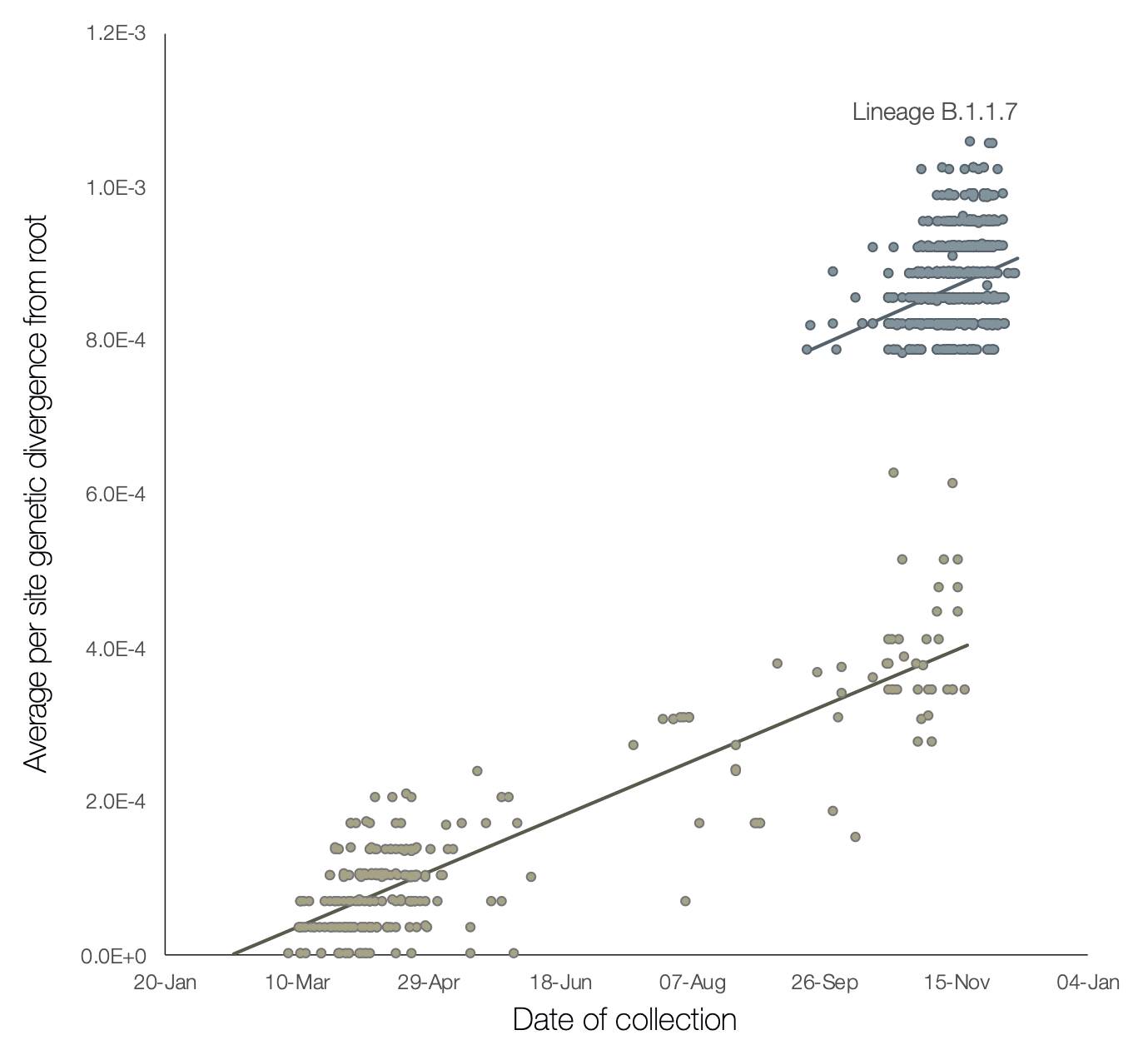
Figure 2 | Regression of root-to-tip genetic distances against sampling dates, for sequences belonging to lineage B.1.1.7 (blue) and those in its immediate outgroup in the global phylogenetic tree (brown). The regression lines are fitted to the two sets independently. The regression gradient is an estimate of the rate of sequence evolution. These rates are 5.6E-4 and 5.3E-4 nucleotide changes/site/year for the B.1.1.7 and outgroup data sets, respectively.
What evolutionary processes or selective pressures might have given rise to lineage B.1.1.7?
High rates of mutation accumulation over short time periods have been reported previously in studies of immunodeficient or immunosuppressed patients who are chronically infected with SARS-CoV-2 (Choi et al. 2020; Avanzato et al. 2020; Kemp et al. 2020). These infections exhibit detectable SARS-CoV-2 RNA for 2-4 months or longer (although there are also reports of long infections in some immunocompetent individuals). The patients are treated with convalescent plasma (sometimes more than once) and usually also with the drug remdesivir. Virus genome sequencing of these infections reveals unusually large numbers of nucleotide changes and deletion mutations and often high ratios of non-synonymous to synonymous changes. Convalescent plasma is often given when patient viral loads are high, and Kemp et al. (2020) report that intra-patient virus genetic diversity increased after plasma treatment was given.
Under such circumstances, the evolutionary dynamics of and selective pressures upon the intra-patient virus population are expected to be very different to those experienced in typical infection. First, selection from natural immune responses in immune-deficient/suppressed patients will be weak or absent. Second, the selection arising from antibody therapy may be strong due to high antibody concentrations. Third, if antibody therapy is administered after many weeks of chronic infection, the virus population may be unusually large and genetically diverse at the time that antibody-mediated selective pressure is applied, creating suitable circumstances for the rapid fixation of multiple virus genetic changes through direct selection and genetic hitchhiking.
These considerations lead us to hypothesise that the unusual genetic divergence of lineage B.1.1.7 may have resulted, at least in part, from virus evolution with a chronically-infected individual. Although such infections are rare, and onward transmission from them presumably even rarer, they are not improbable given the ongoing large number of new infections.
Although we speculate here that chronic infection played a role in the origins of the B.1.1.7 variant, this remains a hypothesis and we cannot yet infer the precise nature of this event.
Potential biological significance of mutations
Table 1 provides details of the B.1.1.7 lineage-specific non-synonymous mutations and deletions. We note that many occur in the virus spike protein. These include spike position 501, one of the key contact residues in the receptor binding domain (RBD), and experimental data suggests mutation N501Y can increase ACE2 receptor affinity (Starr et al. 2020) and P681H, one of 4 residues comprising the insertion that creates a furin cleavage site between S1 and S2 in spike. The S1/S2 furin cleavage site of SARS-CoV-2 is not found in closely related coronaviruses and has been shown to promote entry into respiratory epithelial cells and transmission in animal models (Hoffmann, Kleine-Weber, and Pöhlmann 2020; Peacock et al. 2020; Zhu et al. 2020). N501Y has been associated with increased infectivity and virulence in a mouse model (Gu et al. 2020). Both N501Y and P681H have been observed independently but not to our knowledge in combination before now.
Also present is the deletion of two amino acids at sites 69-70 in spike - this mutation is one of a number of recurrent deletions observed in the N terminal domain of Spike (McCarthy et al. 2020; Kemp et al. 2020) and has been seen in multiple lineages linked to several RBD mutations. For example, it arose in the mink-associated outbreak in Denmark on the background of the Y453F RBD mutation, and in humans in association with the N439K RBD mutation, accounting for its relatively high frequency in the global genome data (~3000 sequences).
Table 1 | Non-synonymous mutations and deletions inferred to occur on the branch leading to lineage B.1.1.7 lineage.
| gene | nucleotide | amino acid |
|---|---|---|
| ORF1ab | C3267T | T1001I |
| C5388A | A1708D | |
| T6954C | I2230T | |
| 11288-11296 deletion | SGF 3675-3677 deletion | |
| spike | 21765-21770 deletion | HV 69-70 deletion |
| 21991-21993 deletion | Y144 deletion | |
| A23063T | N501Y | |
| C23271A | A570D | |
| C23604A | P681H | |
| C23709T | T716I | |
| T24506G | S982A | |
| G24914C | D1118H | |
| Orf8 | C27972T | Q27stop |
| G28048T | R52I | |
| A28111G | Y73C | |
| N | 28280 GAT->CTA | D3L |
| C28977T | S235F |
Outside of spike, the ORF8 Q27stop mutation truncates the ORF8 protein or renders it inactive and thus allows further downstream mutations to accrue. Early on during the pandemic multiple virus isolates with deletions leading to loss of ORF8 expression were isolated worldwide, including a large cluster in Singapore with a deletion leading to both a truncated Orf7b and ablated ORF8 expression. The Singaporean strain, which had a 382nt deletion, was associated with a milder clinical infection and less post-infection inflammation, however this cluster died out at the end of March after Singapore successfully implemented control measures (Young et al. 2020). Subsequent work has found that the ORF8 deletion has only a very modest effect on virus replication in human primary airway cells compared to viruses without the deletion, leading to a slight replication lag compared to viruses with the deletion (Gamage et al. 2020). As ORF8 is usually 121 amino acids long it is likely the stop codon at position 27 observed in lineage B.1.1.7 results in a loss of function.
Finally there are 6 synonymous mutations with 5 in ORF1ab (C913T, C5986T, C14676T, C15279T, T16176C), and one in the M gene (T26801C).
Conclusion
We report a rapidly growing lineage in the UK associated with an unexpectedly large number of genetic changes including in the receptor-binding domain and associated with the furin cleavage site. Given (i) the experimentally-predicted and plausible phenotypic consequences of some of these mutations, (ii) their unknown effects when present in combination, and (iii) the high growth rate of B.1.1.7 in the UK, this novel lineage requires urgent laboratory characterisation and enhanced genomic surveillance worldwide.
References
Avanzato, Victoria A., M. Jeremiah Matson, Stephanie N. Seifert, Rhys Pryce, Brandi N. Williamson, Sarah L. Anzick, Kent Barbian, et al. 2020. “Case Study: Prolonged Infectious SARS-CoV-2 Shedding from an Asymptomatic Immunocompromised Individual with Cancer.” Cell, November. Redirecting.
Choi, Bina, Manish C. Choudhary, James Regan, Jeffrey A. Sparks, Robert F. Padera, Xueting Qiu, Isaac H. Solomon, et al. 2020. “Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host.” The New England Journal of Medicine 383 (23): 2291–93.
Duchene, Sebastian, Leo Featherstone, Melina Haritopoulou-Sinanidou, Andrew Rambaut, Philippe Lemey, and Guy Baele. 2020. “Temporal Signal and the Phylodynamic Threshold of SARS-CoV-2.” Virus Evolution 6 (2): veaa061.
Young, Barnaby E. et al. 2020. “Effects of a Major Deletion in the SARS-CoV-2 Genome on the Severity of Infection and the Inflammatory Response: An Observational Cohort Study.” 2020. The Lancet 396 (10251): 603–11.
Gamage, Akshamal M., Kai Sen Tan, Wharton O. Y. Chan, Jing Liu, Chee Wah Tan, Yew Kwang Ong, Mark Thong, et al. 2020. “Infection of Human Nasal Epithelial Cells with SARS-CoV-2 and a 382-Nt Deletion Isolate Lacking ORF8 Reveals Similar Viral Kinetics and Host Transcriptional Profiles.” PLoS Pathogens 16 (12): e1009130.
Gu, Hongjing, Qi Chen, Guan Yang, Lei He, Hang Fan, Yong-Qiang Deng, Yanxiao Wang, et al. 2020. “Adaptation of SARS-CoV-2 in BALB/c Mice for Testing Vaccine Efficacy.” Science 369 (6511): 1603–7.
Hoffmann, Markus, Hannah Kleine-Weber, and Stefan Pöhlmann. 2020. “A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells.” Molecular Cell 78 (4): 779–84.e5.
Kemp, S. A., D. A. Collier, R. Datir, S. Gayed, A. Jahun, M. Hosmillo, Iatm Ferreira, et al. 2020. “Neutralising Antibodies Drive Spike Mediated SARS-CoV-2 Evasion.” Infectious Diseases (except HIV/AIDS). medRxiv. https://doi.org/10.1101/2020.12.05.20241927
McCarthy, Kevin R., Linda J. Rennick, Sham Nambulli, Lindsey R. Robinson-McCarthy, William G. Bain, Ghady Haidar, and W. Paul Duprex. 2020. “Natural Deletions in the SARS-CoV-2 Spike Glycoprotein Drive Antibody Escape.” Microbiology. bioRxiv.
Peacock, Thomas P., Daniel H. Goldhill, Jie Zhou, Laury Baillon, Rebecca Frise, Olivia C. Swann, Ruthiran Kugathasan, et al. 2020. “The Furin Cleavage Site of SARS-CoV-2 Spike Protein Is a Key Determinant for Transmission due to Enhanced Replication in Airway Cells.” Cold Spring Harbor Laboratory. https://doi.org/10.1101/2020.09.30.318311.
Starr, Tyler N., Allison J. Greaney, Sarah K. Hilton, Daniel Ellis, Katharine H. D. Crawford, Adam S. Dingens, Mary Jane Navarro, et al. 2020. “Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding.” Cell 182 (5): 1295–1310.e20.
Zhu, Yunkai, Fei Feng, Gaowei Hu, Yuyan Wang, Yin Yu, Yuanfei Zhu, Wei Xu, et al. 2020. “The S1/S2 Boundary of SARS-CoV-2 Spike Protein Modulates Cell Entry Pathways and Transmission.” Cold Spring Harbor Laboratory. https://doi.org/10.1101/2020.08.25.266775.