Update of the SARS-CoV-2 genomic surveillance in the Amazonas state, Brazil
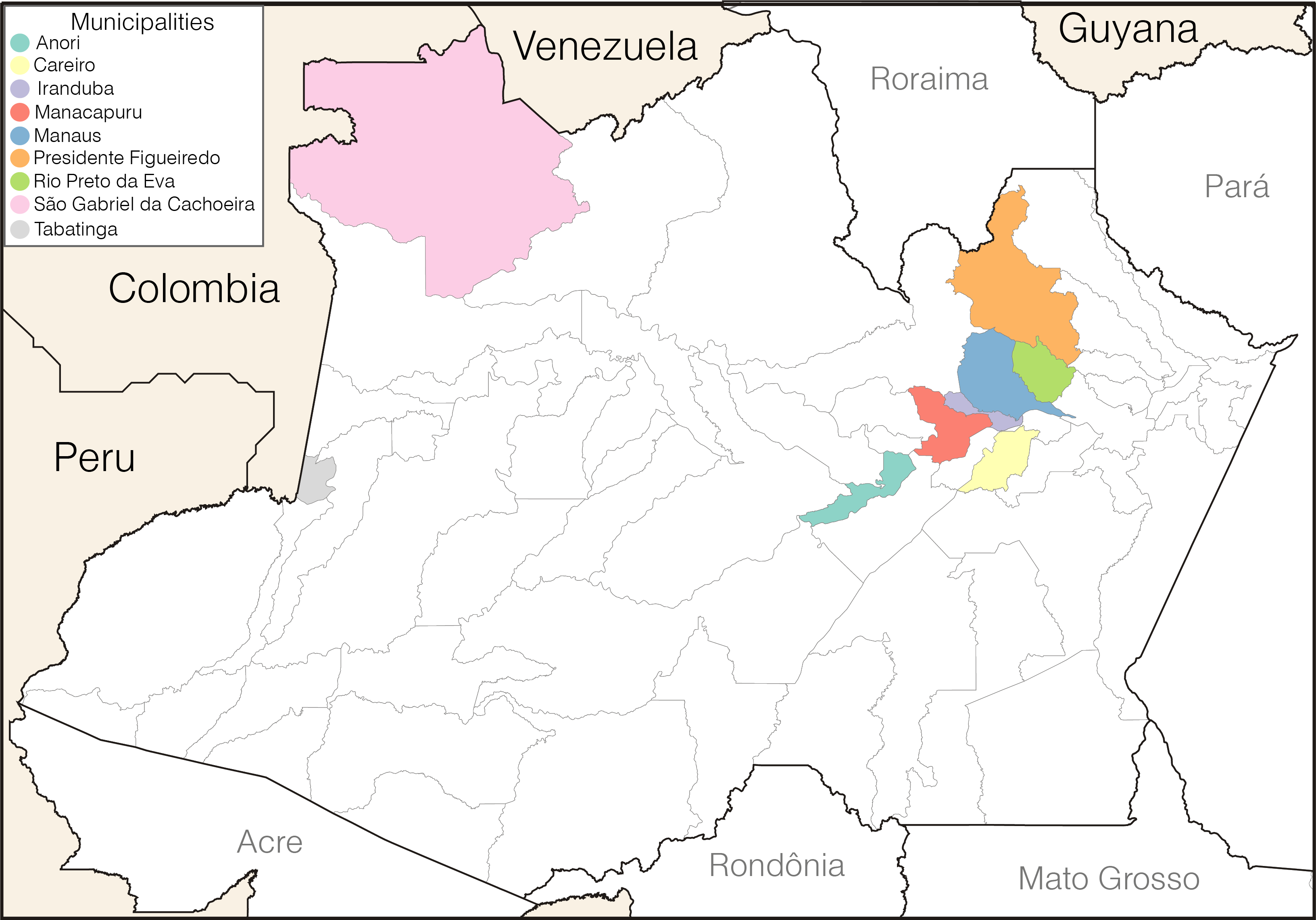
Here follows an update of our previous post showing lineage assignment of 114 SARS-CoV-2 high coverage (zero Ns) and complete (>29.5 Kb) genome sequences collected in different municipalities of the Amazonas state between 1st November 2020 and 13th January 2021. Our data shows the first detection of lineage P.1 on 4th December 2020 in Manaus and its subsequent identification in other municipalities across the Amazonas state. These include not only several municipalities of the Manaus metropolitan region (i.e., Careiro, Iranduba, Manacapuru, Presidente Figueiredo, and Rio Preto da Eva), but also cities located up to 1,100 Km of distance from Manaus and at the border region with Peru, Colombia and Venezuela (São Gabriel da Cachoeira and Tabatinga) (Figure 1). Our data support a rapid increase in the proportion of P.1 cases in the Amazonas state from 0% in November 2020 (n=0/24), to 51% in December 2020 (n=28/55) and 91% in January 2021 (n=32/35) (Table). The P.2 variant, by contrast, was only detected in Manaus at a relatively low and constant frequency (4-11%) between November 2020 and January 2021.
Table. Temporal distribution of SARS-CoV-2 lineages in the Amazonas state.
| Time period | P.1 | P.2 | B.1.1.28 | Others | Total |
|---|---|---|---|---|---|
| November 2020 | 0 | 1 (4%) | 19 (79%) | 4 (17%) | 24 |
| December 2020 | 28 (51%) | 6 (11%) | 17 (31%) | 4 (7%) | 55 |
| January 2021 | 32 (91%) | 2 (6%) | 0 | 1 (3%) | 35 |
Figure 1. Municipalities of the Amazonas state with SARS-Cov-2 P.1 lineage samples sequenced in this study.
A closer inspection of the genetic diversity within the previously defined Amazonian B.1.1.28 clade 28-AM-II revealed a P.1-like sequence sampled in Manaus on 23th December 2020 that branched basal to the P.1 lineage (Figure 2A). This P.1-like sequence harbors 6/10 P.1 lineage-defining mutations in the Spike protein (L18F, P26S, D138Y, K417T, E484K, N501Y) as well as P.1 lineage-defining mutations in the NSP3 (K977Q), NS3 (S253P) and N (P80R) proteins. The P.1-like sequence did not display the nine nucleotides deletion at NSP6 (S106del, G107del, F108del) nor the four nucleotides insertion at position 28269-28273, but displayed a 12 nucleotides insertion at the Spike (ins214ARNR). Similar to the P.1 lineage, the P.1-like sequence also accumulated an unusual high number of genetic changes (Figure 2B). Our sampling effort from November and December in Manaus further revealed a small clade (n = 3) of B.1.1.33 lineage sequences harboring the mutation E484K in the Spike protein.
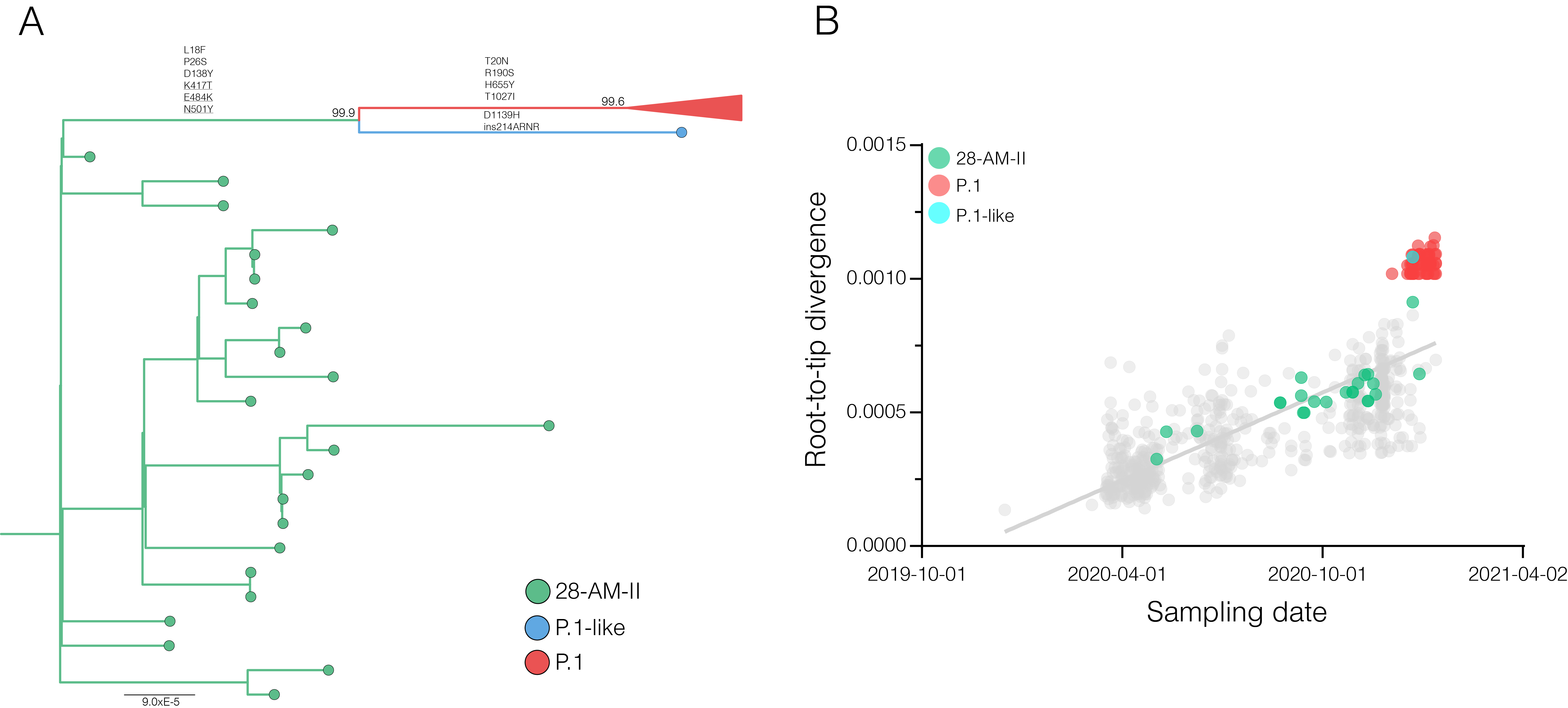
Figure 2. Detection of highly divergent P.1 and P.1-like variants in Manaus. A) Maximum likelihood (ML) phylogenetic tree of the B.1.1.28 whole-genome sequences from the Amazonas state classified within clade 28-AM-II and lineage P.1. The lineage P.1 is collapsed for visual clarity. Branches and tips are coloured according to the clade/lineage as indicated in the legend at the bottom right. The aLRT support values are indicated in key nodes. Non-synonymous lineage-defining mutations in the Spike protein common to both P.1 and P.1-like variants and those specific to each variant are indicated in the corresponding branches. Branch lengths are drawn to scale with the bar indicating nucleotide substitutions per site. B) Correlation between the sampling date of Brazilian B.1.1.28 sequences and their genetic distance from the root of the ML phylogenetic tree. Points corresponding to the clade 28-AM-II, sequence P.1-like and lineage P.1 are colored as indicated in the legend.
These findings clearly support that most SARS-CoV-2 cases detected in Manaus since December 2020 are being caused by the P.1 lineage, in agreement with those described by Faria et al. (https://pando.tools/t/genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-manaus-preliminary-findings/586/2). Our results also point that the P.1 lineage emerged in a short time frame and disseminated fast outside Manaus reaching nearby municipalities of the Metropolitan region as well as very distant municipalities located at the border region with neighboring South American countries. Furthermore, the detection of a P.1-like basal genome, harboring some of the P.1 lineage-defining mutations, and a group of B.1.1.33-E484K viruses in Manaus suggests that the diversity of SARS-CoV-2 variants carrying mutations of concern at the Spike protein circulating in this Brazilian city in late 2020 could be larger than initially described.