Phylogenetic relationship of SARS-CoV-2 sequences from Amazonas with emerging Brazilian variants harboring mutations E484K and N501Y in the Spike protein
Felipe Naveca 1, Valdinete Nascimento 1, Victor Souza 1, André Corado 1, Fernanda Nascimento 1, George Silva 1, Ágatha Costa 1, Débora Duarte 1, Karina Pessoa 1, Luciana Gonçalves 2, Maria Júlia Brandão 1, Michele Jesus 3, Cristiano Fernandes 2, Rosemary Pinto 2, Marineide Silva 4, Tirza Mattos 4, Gabriel Luz Wallau 5, Marilda Mendonça Siqueira 6, Paola Cristina Resende 6*, Edson Delatorre 7*, Tiago Gräf 8*, Gonzalo Bello 9*
*These authors contributed equally to this work
1 Laboratório de Ecologia de Doenças Transmissíveis na Amazônia, Instituto Leônidas e Maria Deane, Manaus, Amazonas, Brazil.
2 Fundação de Vigilância em Saúde do Amazonas, Manaus, Amazonas, Brazil.
3 Laboratório de Diversidade Microbiana da Amazônia com Importância para a Saúde, Instituto Leônidas e Maria Deane, Manaus, Amazonas, Brazil.
4 Laboratório Central de Saúde Pública do Amazonas, Manaus, Amazonas, Brazil.
5 Instituto Aggeu Magalhães, Fundação Oswaldo Cruz, Recife, Pernambuco, Brazil.
6 Laboratory of Respiratory Viruses and Measles, Oswaldo Cruz Institute (IOC), Oswaldo Cruz Foundation (FIOCRUZ), Rio de Janeiro, Brazil. SARS-CoV-2 National Reference Laboratory for the Brazilian Ministry of Health (MoH) and Reference Laboratory for the World Health Organization (WHO).
7 Departamento de Biologia. Centro de Ciências Exatas, Naturais e da Saúde, Universidade Federal do Espírito Santo, Alegre, Brazil.
8 Instituto Gonçalo Moniz, Fundação Oswaldo Cruz, Salvador, Brazil.
9 Laboratório de AIDS e Imunologia Molecular, Instituto Oswaldo Cruz, FIOCRUZ, Rio de Janeiro, 40 Brazil.
Summary
Here we report a preliminary genomic analysis of SARS-CoV-2 B.1.1.28 lineage circulating in the Brazilian Amazon region and their evolutionary relationship with emerging and potential emerging SARS-CoV-2 Brazilian variants harboring mutations in the RBD of Spike (S) protein. Phylogenetic analysis of 69 B.1.1.28 sequences isolated in the Amazonas state revealed the existence of two major clades that have evolved locally without unusual mutations in the S protein from April to November 2020. The B.1.1.28 viruses harboring mutations S:K417N, S:E484K and S:N501Y, recently detected in Japanese travelers returning from Amazonas, branched within one of the Amazonian B.1.1.28 clades here identified, suggesting that these sequences could be representatives of a novel (unreported) emerging Brazilian clade, here designated B.1.1.28(K417N/E484K/N501Y). Our analysis also confirms that the putative novel clade B.1.1.28(K417N/E484K/N501Y) detected in Japanese travelers did not evolve from the clade B.1.1.28(E484K) recently detected in Rio de Janeiro and other Brazilian states, but both variants arose independently during the evolution of the B.1.1.28 lineage.
Background
The emergence of new SARS-CoV-2 variants harboring mutations in the Spike protein that might impact viral fitness and transmissibility has been an issue of great concern, particularly after the recent identification of two independent emerging strains in UK and South Africa with a larger than usual number of mutations in the Spike protein that may have functional significance. Both the emerging SARS-CoV-2 lineages B.1.1.7 in UK (HV 69-70 deletion, Y144 deletion, N501Y, A570D, P681H, T716I, S982A, D1118H) (1) and B.1.351 in South Africa (L18F, D80A, D215G, R246I, K417N, E484K, N501Y and A701V) (2) acquired eight lineage-defining amino acid replacements in the Spike protein, being mutation N501Y in the receptor-binding domain (RBD) the only common amino acid replacement detected in both lineages.
The SARS-CoV-2 epidemic in Brazil was dominated by two lineages designated as B.1.1.28 and B.1.1.33 that probably emerged in the country in February 2020 (3,4). Recent reports bring attention to the emergence of new SARS-CoV-2 B.1.1.28 variants in Brazil harboring mutations in the Spike protein common to the B.1.1.7 and B.1.351 lineages. A study published in December 2020 described the emergence of a novel B.1.1.28 clade in the state of Rio de Janeiro that was distinguished by five lineage-defining mutations, including one in the Spike protein (E484K), also detected in the B.1.351 South African lineage (5), but of independent origin. This clade, here designated as B.1.1.28(E484K), was subsequently detected in other Brazilian states and was further associated with two cases of reinfection in patients originally infected by the B.1.1.33 lineage (6). More recently, the Japanese Ministry of Health reported the presence of a new SARS-CoV-2 B.1.1.28 variant in four travelers that arrived in Japan returning from Amazonas state (northern Brazil) on 2nd January 2021 that harbors 10 synapomorphic mutations in the Spike (L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, H655Y, T1027I), including one (N501Y) detected in both the B.1.351 and the B.1.1.7 lineages and three also detected in the B.1.351 South African lineage only (L18F, K417N, E484K) (7).
In order to have a more in depth understanding of the origin of the new B.1.1.28 variant detected in Japan, here designated as B.1.1.28(K417N/E484K/N501Y), we analyzed the genetic diversity of 148 SARS-CoV-2 whole-genome strains circulating in the Brazilian Amazon state between April and November, 2020.
Results
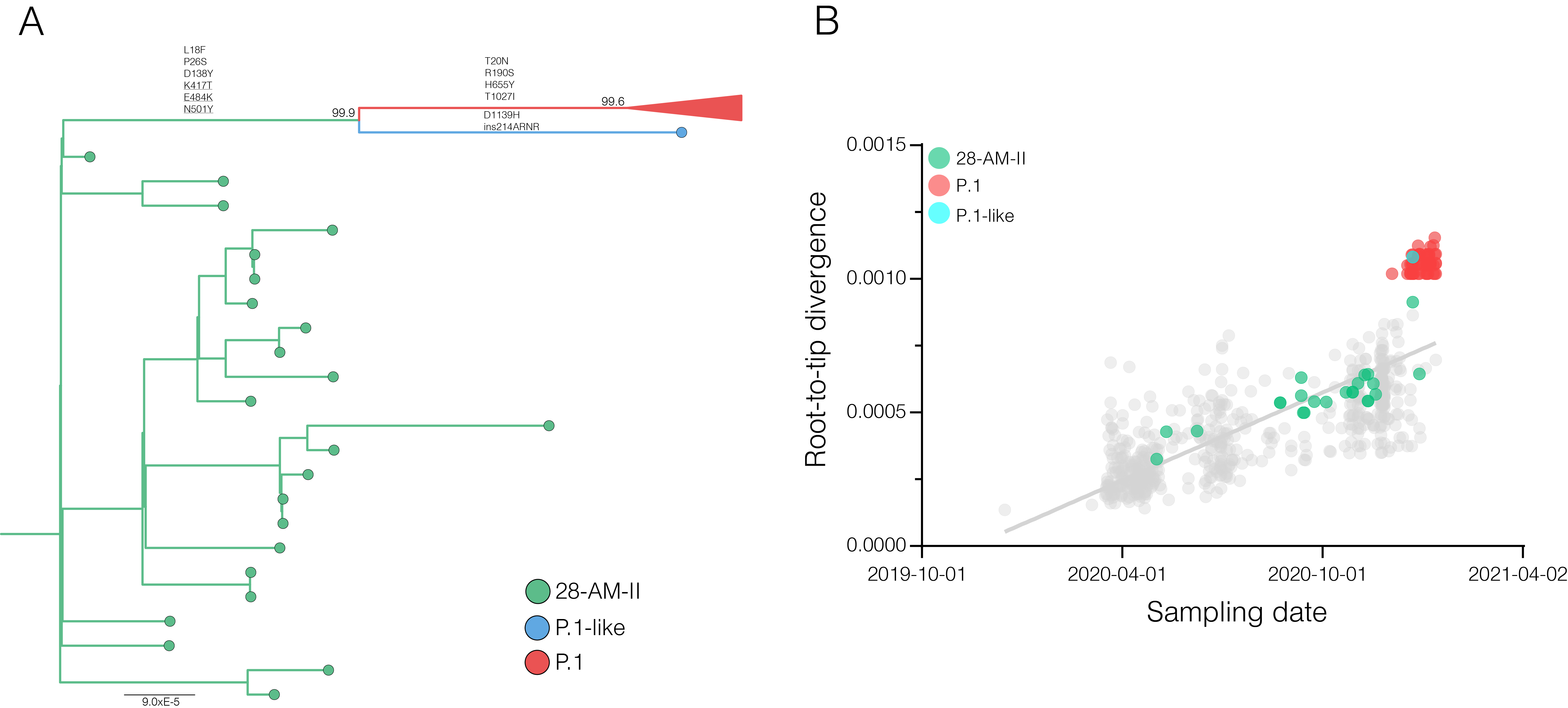
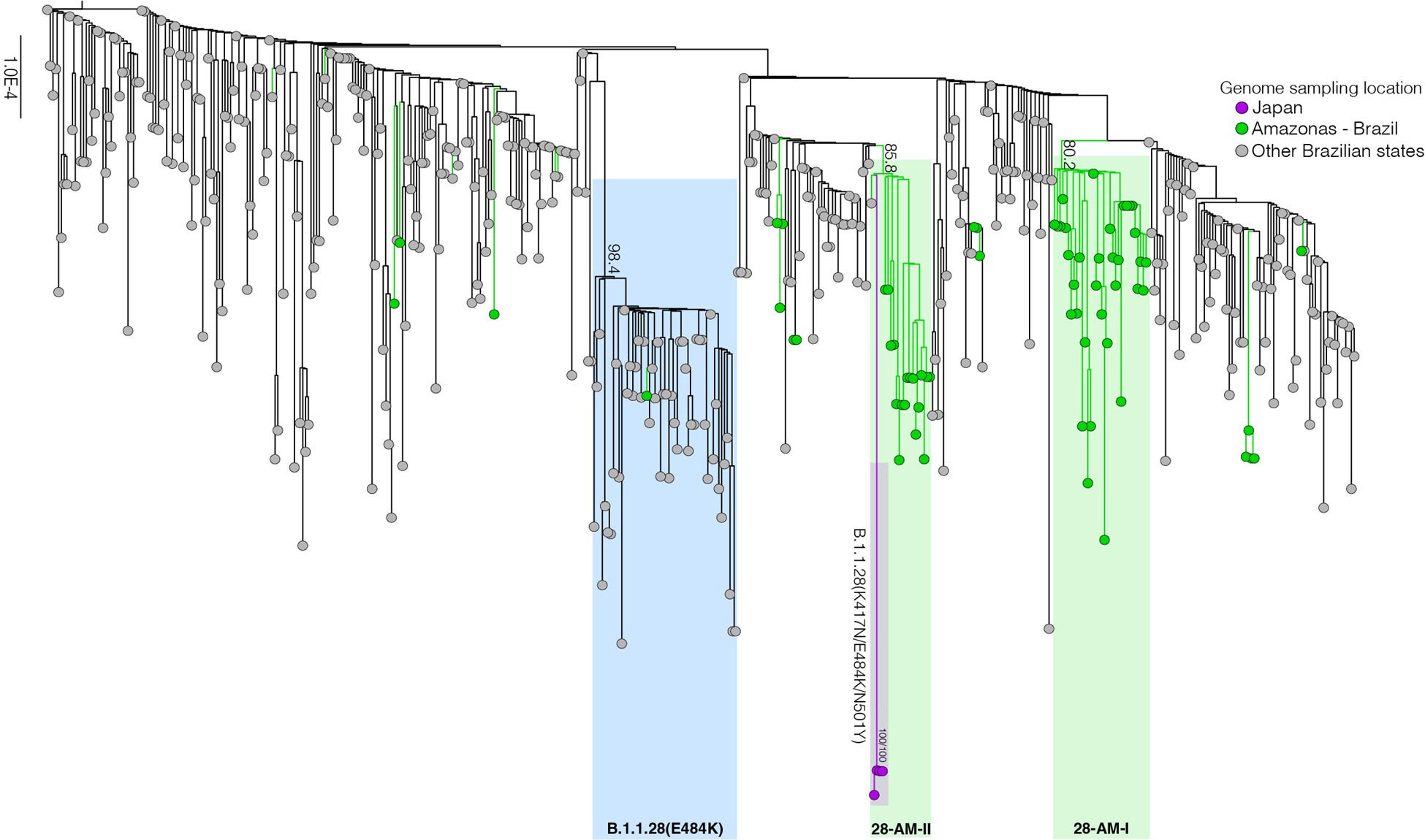
Our genomic survey of 148 SARS-CoV-2 whole-genome sequences from Amazonas state, identified 69 (47%) B.1.1.28 sequences sampled from different municipalities between 13th April and 13th November, 2020, being the most prevalent viral lineage in this Brazilian state (Figure 1). These B.1.1.28 sequences were next aligned with 399 B.1.1.28 high-quality (<10% of N) whole-genome (>29.000 nucleotides long) sequences of Brazilian origin and the four B.1.1.28 sequences from Japanese travelers returning from Amazonas that were available in the EpiCoV database in GISAID by 10th January, 2021. Maximum-likelihood (ML) phylogenetic analyses using IQTree v2.1.2 8 revealed that Brazilian B.1.1.28 sequences from Amazonas were mostly branched in two highly supported (approximate likelihood-ratio test [aLRT] > 80%) monophyletic clades here designated as 28-AM-I (49%) and 28-AM-II (26%) (Figure 2). The monophyletic clade 28-AM-I comprises 34 sequences isolated in different Amazonian municipalities between 20th April and 13th November that were nested among basal sequences from the Sao Paulo state. The monophyletic clade 28-AM-II comprises 18 sequences isolated in different Amazonian municipalities between 20th April and 13th November (EPI_ISL_801386 to EPI_ISL_801403) and also comprises the four Japanese B.1.1.28 sequences sampled from travelers returning from the Amazon region. The clade 28-AM-II was nested within a basal paraphyletic clade (aLRT = 77%) that comprises sequences sampled from different Brazilian states between 1st April and 11th November, 2020 (Figure 2). The emerging B.1.1.28(E484K) Brazilian lineage appeared as an independent highly supported monophyletic clade (aLRT = 98%) within the B.1.1.28 phylogeny and comprises sequences from different Brazilian states, including the only B.1.1.28 sequence from the Amazon state harboring the E484K mutation detected in our study (Figure 2). Bayesian reconstruction using a strict molecular clock model with a normal substitution rate prior (8-10 × 10-4 substitutions/site/year) as implemented in BEAST 1.10 9 estimated the emergence of the clade 28-AM-II at 27th April (95% High Posterior Density [HPD]: 30th March – 28th April).
Figure 1. Geographic and temporal distribution of SARS-CoV-2 B.1.1.28 sequences sampled in the Amazonas state. A) Municipalities of the Amazonas state with SARS-Cov-2 B.1.1.28 samples sequenced in this study. B) SARS-CoV-2 B.1.1.28 sequences obtained in this study (black dots) and confirmed SARS-CoV-2 cases (gray bars) in Amazonas by epidemiological week (EW).
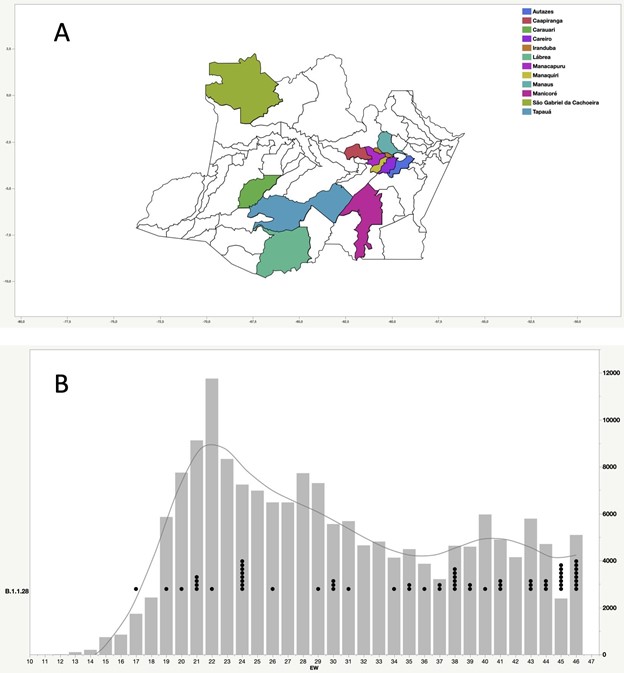
Figure 2. Maximum likelihood (ML) phylogenetic tree of the B.1.1.28 whole-genome sequences from Brazil and Japan. The sequences from Amazonas state are represented by green circles and those sampled in Japan by the purple ones. The B.1.1.28(E484K) cluster is highlighted in blue, while the two clusters identified in the Amazonas state (28-AM-I and 28-AM-II) are highlighted in green. The aLRT support values are indicated in key nodes. The tree was rooted in the oldest sample and branch lengths are drawn to scale with the left bar indicating nucleotide substitutions per site.
Most B.1.1.28 Brazilian clades identified so far were characterized by a low number of synapomorphic mutations that distinguish them from other Brazilian B.1.1.28 sequences. We identify one lineage-defining synonymous mutation in clade 28-AM-I (C29284T), two mutations in clade 28-AM-II and basal sequences (A6319G and T26149C [ORF3a:S253P]), one synonymous mutation exclusive of clade 28-AM-II (A6613G), and five mutations in clade B.1.1.28(E484K) (C100U [5’UTR], T10667G [NSP5:L205V], C11824T [NSP6], G23012A [S:E484K], and G28628T [N:A119S]) (Table). The pattern is consistent with the accumulation of mutations at a relatively constant rate over time during local evolution and diversification of lineage B.1.1.28 in Brazil (Figure 3). The branch leading to the B.1.1.28 sequences sampled from Japanese travelers, by contrast, accumulated an unusual high number of genetic changes (Figure 3), in addition to those in the Spike protein (Table), which resemble the pattern observed in the B.1.1.7 and B.1.351 lineages from UK and South Africa. None of the B.1.1.28 sequences from Amazonas detected in our genomic survey displayed such a divergent pattern of nucleotide and amino acid substitutions.
Table. Lineage-defining mutations of the different B.1.1.28 clades detected in Brazil and Japan. Shaded boxes highlight the B.1.1.28 lineage-defining mutation (dark gray) and those common to clades B.1.1.28-AM-II and B.1.1.28(K417T/E484K/N501Y) (light gray).
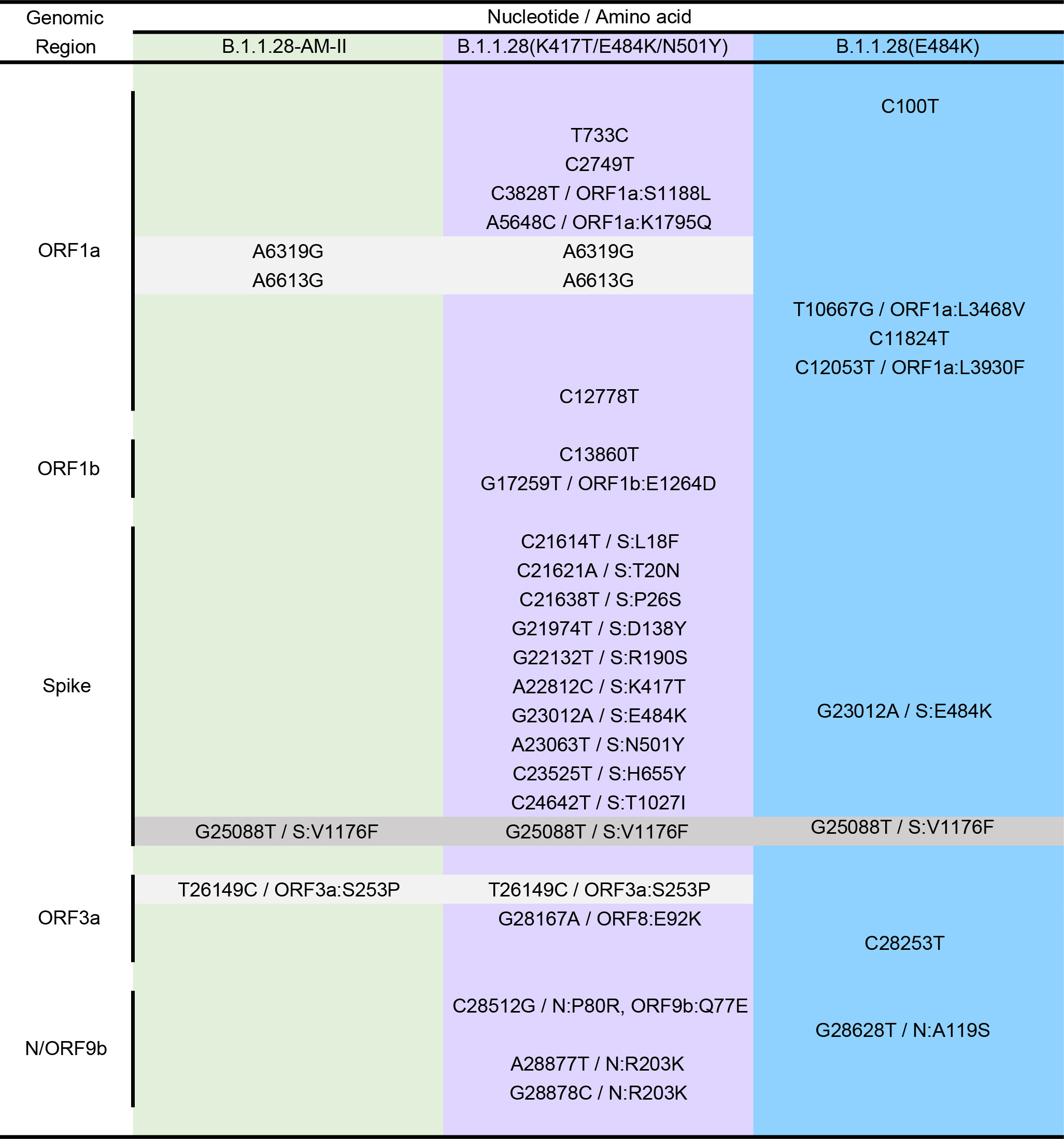
Figure 3. Correlation between the sampling date of B.1.1.28 sequences and their genetic distance from the root of the ML phylogenetic tree. Colours indicate the B.1.1.28 clade of the corresponding sequences according to the legend at left.
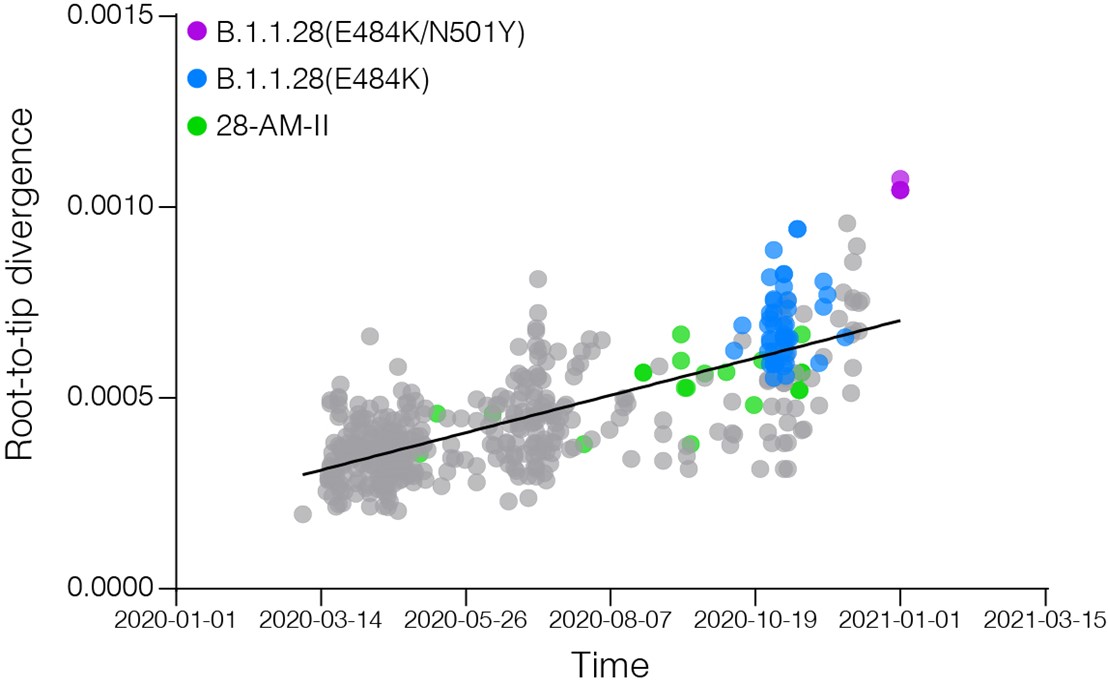
These findings support that the SARS-CoV-2 B.1.1.28 strains detected in Japanese travelers returning from the Brazilian Amazon region probably evolved from a viral lineage that circulates in the Amazonas state since April 2020, and might be representatives of a potentially new emerging Brazilian lineage, here designated as B.1.1.28(K417N/E484K/N501Y). Local B.1.1.28 Amazonian lineages seem to have evolved at a constant rate between April and November 2020 and none of the sequences here obtained displayed an unusually high number of mutations in the Spike or in any other genomic region. These findings support that the fast mutation rate detected in the B.1.1.28(K417N/E484K/N501Y) variant is a recent phenomena that probably took place between December 2020 - January 2021. Aiming to detect the circulation of this lineage among the Amazonian population, we are currently conducting a genomic survey of SARS-CoV-2 recently infected individuals in Amazonas . Our preliminary analysis also confirms that the emerging Brazilian lineages B.1.1.28(E484K) and B.1.1.28(K417N/E484K/N501Y) arose independently during the diversification of the B.1.1.28 lineage in Brazil. The concurrent emergence of different viral B.1.1 lineages carrying mutations K417N, E484K and N501Y in the RBD of the Spike protein in different countries around the world during the second half of 2020 suggests convergent selective changes in SARS-CoV-2 evolution due to similar evolutionary pressure during the process of infection of millions of people. If these mutations confer some selective advantage for the viral transmissibility we should expect an increasing frequency of those viral lineages in Brazil and around the world in the following months.
Acknowledgments
The authors wish to thank all the health care workers and scientists who have worked hard to deal with this pandemic threat, the GISAID team, and all the EpiCoV database’s submitters, in particular the Japanese National Institute of Infectious Diseases (NIID) members Dr. Tsuyoshi Sekizuka, Dr. Kentaro Itokawa, Rina Tanaka and Masanori Hashino to publish the genomes. GISAID acknowledgment table containing sequences used in this study are attached to this post (Supplementary Table 1). We would also wish to thank Dr Nuno Faria from https://www.caddecentre.org for sharing their unpublished findings regarding the SARS-CoV-2 B.1.1.28 lineage. We also appreciate the support of Genomic Coronavirus Fiocruz Network members and the Respiratory Viruses Genomic Surveillance Network of the General Laboratory Coordination (CGLab) of the Brazilian Ministry of Health (MoH), Brazilian Central Laboratory States (LACENs), and the Amazonas surveillance teams for the partnership in the viral surveillance in Brazil. Funding support FAPEAM (PCTI-EmergeSaude/AM call 005/2020 and Rede Genômica de Vigilancia em Saúde - REGESAM); Conselho Nacional de Desenvolvimento Científico e Tecnológico (grant 403276/2020-9); Inova Fiocruz/Fundação Oswaldo Cruz (Grant VPPCB-007-FIO-18-2-30 - Geração de conhecimento).
Suplemetary Table 1. Suplementary_table 1_acknowledgement_table_GISAID.pdf (47.3 KB)
References
1 Rambaut, A. L., N.; Pybus, O.; Barclay, W.; Barrett, J.; Carabelli, A.; Connor, T.; Peacock, T.; Robertson, D.; Volz, E.; on behalf of COVID-19 Genomics Consortium UK (CoG-UK); . Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. Virological.org (2020). https://pando.tools/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563.
2 Tegally, H. et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv, doi:10.1101/2020.12.21.20248640 (2020).
3 Candido, D. S. et al. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science 369, 1255-1260, doi:10.1126/science.abd2161 (2020).
4 Resende P. C., D., E., Gräf T., Mir D., Motta F.C., Appolinario L., Paixão A. C., Mendonça A. C., Ogrzewalska M., Caetano B., Wallau G. L., Docena C., Santos M. C., Ferreirra J., Sousa Junior E., Silva S., Fernandes S., Vianna L. A., Souza L., Ferro J. F, Nardy V., Santos C., Riediger I., Debur M., Croda J., Oliveira, W, Abreu A, Bello G… Siqueira M. M. Evolutionary dynamics and dissemination pattern of the SARS-CoV-2 lineage B.1.1.33 during the early pandemic phase in Brazil. Frontier in Microbiology, doi:10.3389/fmicb.2020.615280 (2020).
5 Voloch, C. M. et al. Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. medRxiv, doi:10.1101/2020.12.23.20248598 (2020).
6 Resende, P. C. B., J.F.; Vasconcelos, R.H.T.; Arantes I.; Appolinario L.; Mendonça, A.C.; Paixao, A.C.; Rodrigues A.C.; Silva, T.; Rocha, A.S.; Pauvolid-Corrêa, A.; Motta, F.C.; Teixeira, D.L.F.T.; Carneiro, T.F.O.; Freire Neto, F.P.F.; Herbster, I.D.; Leite, A.B.; Riediger, I.N.; Debur, M.C.; Naveca, F.G.; Almeida, W.; Livorati, M.; Bello, G.; Siqueira, M.M. Spike E484K mutation in the first SARS-CoV-2 reinfection case confirmed in Brazil, 2020. Virological.org (2020). https://pando.tools/t/spike-e484k-mutation-in-the-first-sars-cov-2-reinfection-case-confirmed-in-brazil-2020/584.
7 Japan;, N. I. o. I. D. in Coronavirus disease (COVID-19) 4 (2021).
8 Minh, B. Q. et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol 37, 1530-1534, doi:10.1093/molbev/msaa015 (2020).
9 Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol 4, vey016, doi:10.1093/ve/vey016 (2018).