Phylogenetic evidence that B.1.1.7 has been circulating in the United States since early- to mid-November
Brendan B. Larsen,1, Michael Worobey1*
1Department of Ecology and Evolutionary Biology, University of Arizona
*Corresponding author’s email: [email protected]
Introduction:
When SARS-CoV-2 emerged in China late December 2019 and early January 2020, there was little circulating genetic variation as it was exported across the world. This made estimating the number and timing of introductions difficult1,2. Since then, the virus has accumulated mutations (as all RNA viruses do) in a (largely) clock-like manner, diversifying into a myriad of lineages. One of these lineages, ‘B.1.1.7’, first arose in the UK in September after an episode of heightened mutation, likely within a single host3. This lineage has continued to evolve over the last several months while rapidly increasing in frequency across southeast England and elsewhere. Estimates of its increased transmissibility and a suite of mutations in the spike gene have made this variant a top focus of the scientific community since it was first reported in December 20204,5. The rapid increase in the UK has made export increasingly likely, and, as of 1/17/2021, the lineage has been detected in 55 countries, including the United States6.
The increased sequence variation combined with more comprehensive genome sequencing now compared to during the beginning of pandemic may permit clearer estimates of the number and timing of introductions of SARS-CoV-2. Furthermore, given the increased transmission rate of B.1.1.7 and its grave public health consequences, it is critical to understand how long this lineage has been circulating in the United States, which currently has the highest reported death toll from COVID-19 in the world. The first evidence of this lineage in the US was reported in Colorado on 29 December, 20207. Here, we investigate the dynamics of B.1.1.7 in the US using a phylogenetic approach to (1) compare the importance of independent introductions of the lineage from abroad to domestic circulation within the US and (2) characterize the pace of its early spread within the US. Crucial to the latter goal are estimates of the time of the most recent common ancestors (TMRCAs) of any apparent within-US circulating clades of B.1.1.7. We hope that insights into this viral lineage’s spread so far may also provide clues to its possible future dynamics in the US.
Methods:
We focused on a unique set of US B.1.1.7 genomes sequenced by a partnership between the Centers for Disease Control (CDC), the consumer genomics company Helix, and the sequencing company Illumina who have been investigating so-called ‘S dropout’ or spike gene target failure (SGTF) SARS-CoV-2 test samples from across the United States8,9. Such samples have yielded numerous B.1.1.7 genomes; this lineage contains a deletion at the positions that code for sites 69 and 70 of the S protein, causing qPCR assays that target this region to fail. We wish to recognize the importance of the CDC/Helix/Illumina partnership for these public health and genome surveillance efforts across the United States and gratefully acknowledge their willingness to share the data that made this study possible.
Briefly, we downloaded all European B.1.1.7 sequences with a non-ambiguous sampling date from GISAID as of Jan. 15th and selected a subset of context sequences. These context sequences were chosen to span the entire B.1.1.7 sampling period from September 2020 through the end of December 2020 (the most recent sampling date of the US genomes in our data set) as well as to cover the breadth of diversity that has accumulated. In total we used 145 European (mostly UK) B.1.1.7 genomes as context sequences. After alignment with MAFFT10, we manually inspected alignment for obvious sequencing errors. Three nucleotides (positions 28,280-28,282) were changed to Ns in the US sequences EPI_ISL_802601 and EPI_ISL_802634. These three base pairs were likely sequencing/assembly error since all B.1.1.7 sequences have different nucleotides for that stretch of sequence and the remaining US sequences all contained ‘N’s at those positions. We inferred phylogenies in BEAST11 using a GTR+G substitution model with a strict molecular clock using a normally distributed prior with a mean of 8x10-4 substitutions/site/year and a standard deviation of 5x10-5. A Skygrid12 coalescent tree prior was used with 25 grid points. BEAST analyses were run for 50 million generations with sampling every 2000 and discarding the first 5 million steps as burn-in. All parameters were checked using Tracer to ensure mixing and ESS values were >200. Maximum clade credibility phylogenies were made with TreeAnnotator and visualized with BALTIC13. BALTIC was also used to make a tree with low confidence nodes collapsed with the collapseBranches function included as part of the package.
Results and Discussion:
Among these 50 B.1.1.7 genomes from the United States, sampled up to the end of December 2020, there are at least 5 distinct introductions from abroad (Figure 1). This number is obviously a severe underestimate of the total number of introductions of B.1.1.7 to the US given the small fraction of US SARS-CoV-2 cases that are sequenced, and the use of only these CDC/Helix/Illumina data. Of note, there are additional B.1.1.7 genomes available on GISAID from the United States; however, we used only CDC/Helix/Illumina genomes in part to enable comparisons between (1) B.1.1.7 dynamics in the US as ascertained from this single source of data and (2) B.1.1.7 dynamics in the UK (see below).
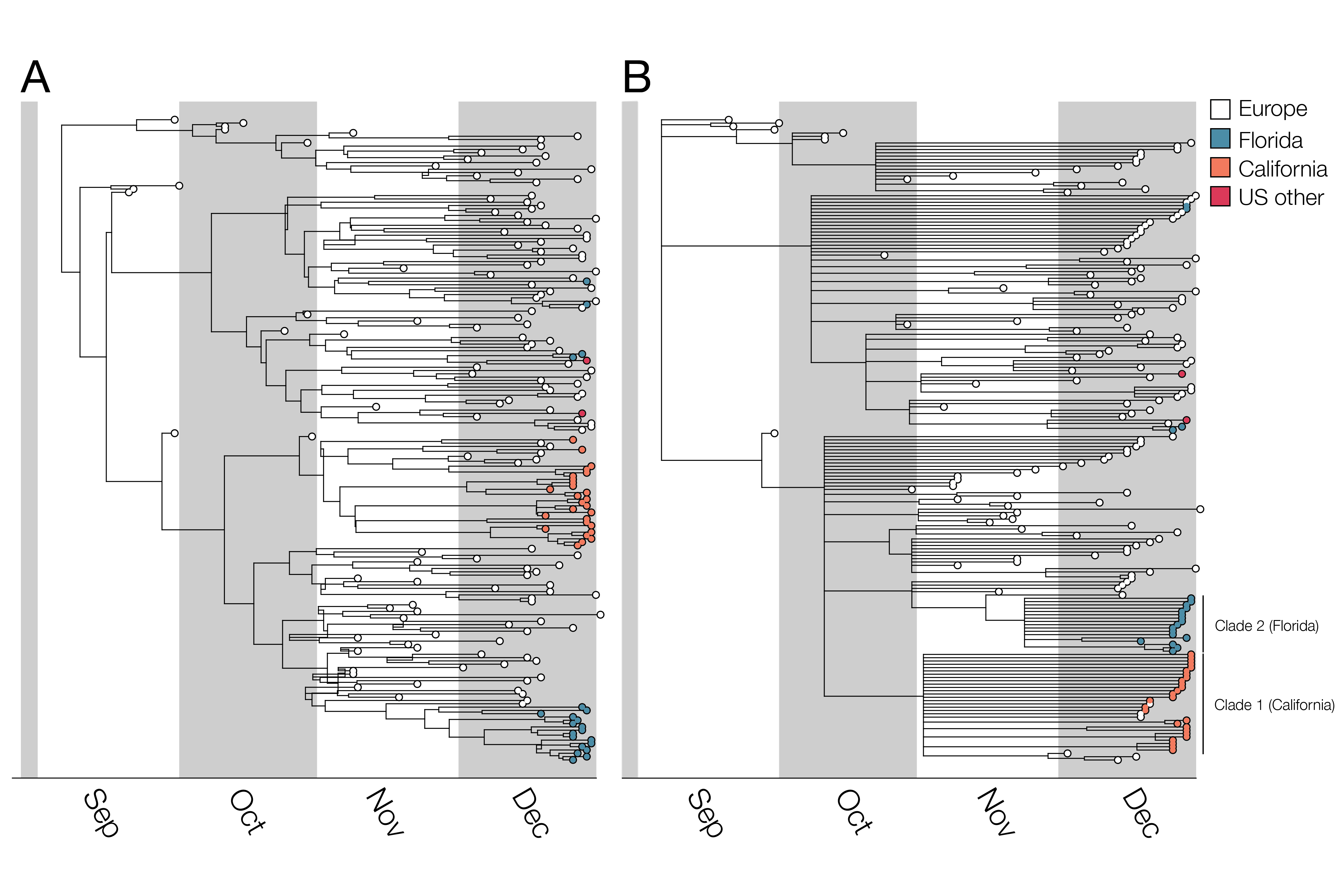
Figure 1. Bayesian phylogenetic analysis of B.1.1.7. A) Maximum clade credibility (MCC) tree and B) MCC tree with nodes with <0.5 posterior probability collapsed to visualize higher confidence lineages. Trees visualized with BALTIC. In the case of the left hand tree there are some internal branches with negative branch lengths. This occurs in certain cases for MCC trees due to low frequencies of particular clades when summarizing over many trees. This phenomenon does not impact our findings and we chose to display B) specifically to only show high confidence phylogenetic placements.
For the two largest clades of B.1.1.7 in the United States, we infer separate introductions of B.1.1.7 into California and Florida, with median TMRCAs of November 6 and November 23, respectively (see Table 1 for highest probability distributions). All clade 1 California sequences share a mutation (G26730C,M:V70L) which is found in a small fraction of European B.1.1.7 sequences (157/12,728; 1.2%). Thus, although some non-US B.1.1.7 viruses intermingle with the California ones due to low phylogenetic signal (Figure 1), the possibility of multiple introductions of this rare genotype (but no additional introductions into California, in this sample, from elsewhere across the tree) is low. Furthermore, all of the Florida sequences in clade 2 differ from the most closely related UK sequence by one mutation (C15720T), a strong indication that they too descend from a single introduction event. The remaining introductions include a group of three viruses from Florida and Georgia, one from Pennsylvania, and two additional genomes from Florida that may or may not descend from a single introduction event. Note that California and Florida are the states for which the most SGTF samples have been sequenced by CDC/Helix/Illumina.
Table1. Median TMRCA and 95% HPD estimates for the two US clades along with the root of B.1.1.7
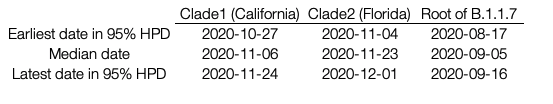
After nearly two months of circulation of a B.1.1.7 lineage in California, the California lineage was estimated to account for a low proportion of cases in Helix data from the state: 0.4% as of December 27 through January 214, some of which might fall outside clade 1 as defined here (December 27 is 51 days from the median TMRCA of clade 1, at November 6). This suggests the dynamics of B.1.1.7 might be somewhat less explosive in California versus its original epicenter in England, with a similar population of 55 million compared to California’s 39 million: when England was at a comparable point into its B.1.1.7 outbreak, B.1.1.7 accounted for approximately 1.2% of SARS-CoV-2 cases, as estimated using GISAID data collected in the week starting 51 days after the September 5 median TMRCA estimate of the B.1.1.7 clade estimated here (Figure 1, Table 1). For this comparison we considered only B.1.1.7 genomes submitted prior to December 9 to exclude a potentially disproportionate number B.1.1.7 genomes sequenced in England after its importance became clear in mid-December.
Clade 2 in Florida (population 21 million), on the other hand, exhibited more rapid displacement of non-B.1.1.7, at least as indicated by this rough comparison approach: 34 days after the Florida clade 2 TMRCA, B.1.1.7 accounted for an estimated 0.7% of cases there14. Since only 17 of 21 (81%) of Florida B.1.1.7 genomes fall into clade 1 (Figure 1), we can further estimate that Clade 1 accounted for approximately 0.6% of all SARS-CoV-2 cases in Florida as of December 27 through January 2. At a comparable point into the B.1.1.7 outbreak in England B.1.1.7 accounted for about 0.1% of all cases there (4 out of 4135 genomes collected the week starting October 8), using the same approach as above. Hence, while it is evidently younger than the California clade 1 lineage, the Florida clade 2 lineage already accounts for a larger proportion of the Florida SARS-CoV-2 epidemic than clade 1 does of the California SARS-CoV-2 outbreak.
It is not clear why the pace of replacement of non-B.1.1.7 viruses might be different in California, Florida and England. We speculate on a few possibilities that need to be monitored as more data become available. One possibility is that B.1.1.7’s transmission advantage may vary with mitigation intensity. Perhaps this lineage of SARS-CoV-2, with demonstrably higher viral loads in the upper airway than other variants4, is able to seed superspreader events with relative ease when mitigation efforts are comparatively lax, but its transmission advantage is less acute when the playing field is leveled by, for example, widespread mask use and indoor crowd avoidance. Another possibility is that the non-B.1.1.7 lineages circulating in the US, particularly in California, may be more transmissible than the non-B.1.1.7 lineages in England with which B.1.1.7 has been competing, giving B.1.1.7 less of a transmission advantage and, thus, a slower displacement rate of non-B.1.1.7 lineages. An example of this might be a new variant with the Spike RBD mutation L452R that was recently described in the press in California15. However, we caution that these preliminary estimates may be very noisy; it will be important to reassess these early apparent trends with additional time points as these outbreaks progress.
It is striking that this lineage may already have been established in the US for some 5-6 weeks before B.1.1.7 was first identified as a variant of concern in the UK in mid-December16. And it may have been circulating in the US for close to 2 months before it was first detected, on 29 December 2020. B.1.1.7 viruses have been estimated to account for only 0.3% of SARS-CoV-2 cases nationally as of early January14. It is also worth noting the vast majority (>90%) of US B.1.1.7 cases appear to be generated in these well-established domestic outbreaks rather than via travel-related introductions from the UK or other affected countries, though such cases are surely also happening.
These results highlight the importance of a global perspective on genome sequencing to detect and monitor new SARS-CoV-2 variants. Given how rapidly new variants can spread across the globe due to air travel, and how long even variants with reportedly increased transmission rates can remain undetected after becoming established in new regions, it is essential that all countries continue efforts to reduce transmission.
Acknowledgements:
We gratefully acknowledge the laboratories and researchers who made all the B.1.1.7 SARS-CoV-2 genomes used in this study available on GISAID, including the CoG-UK consortium (https://www.cogconsortium.uk), who not only first detected the B.1.1.7 lineage17 but also generated most of the context genomes, without which this analysis could not have been done. We also thank Duncan MacCannell for insightful discussions about the CDC/Helix/Illumina B.1.1.7 data and our results.
Acknowledgement table available for download
TableS1_GISAID_acknowledgements.tsv.zip (4.6 KB)
References
- Worobey, M. et al. The emergence of SARS-CoV-2 in Europe and North America. Science 370, 564–570 (2020).
- Bedford, T. et al. Cryptic transmission of SARS-CoV-2 in Washington state. Science 370, 571–575 (2020).
- Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. https://pando.tools/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (2020).
- Kidd, M. et al. S-variant SARS-CoV-2 is associated with significantly higher viral loads in samples tested by ThermoFisher TaqPath RT-QPCR. bioRxiv (2020) doi:10.1101/2020.12.24.20248834.
- Volz, E. et al. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. bioRxiv (2021) doi:10.1101/2020.12.30.20249034.
- SARS-CoV-2 lineages. Redirecting….
- Zimmer, C. & Pietsch, B. First U.S. Case of Highly Contagious Coronavirus Variant Is Found in Colorado. The New York Times (2020).
- Washington, N. L. et al. S gene dropout patterns in SARS-CoV-2 tests suggest spread of the H69del/V70del mutation in the US. bioRxiv (2020) doi:10.1101/2020.12.24.20248814.
- Han, A. Illumina, Helix Collaborate on CDC-Coordinated Coronavirus Surveillance. https://www.genomeweb.com/infectious-disease/illumina-helix-collaborate-cdc-coordinated-coronavirus-surveillance#.YAYmi5NKjRY (2021).
- Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
- Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol 4, vey016 (2018).
- Gill, M. S. et al. Improving Bayesian population dynamics inference: a coalescent-based model for multiple loci. Mol. Biol. Evol. 30, 713–724 (2013).
- Dudas, G. baltic. (Github).
- Update on the Helix, Illumina surveillance program: B.1.1.7 variant of SARS-CoV-2, first identified in the UK, spreads further into the US. https://blog.helix.com/b117-variant-updated-data/ (2021).
- Morris, J. D. New coronavirus variant found in Bay Area linked to massive Kaiser outbreak. https://www.sfchronicle.com/bayarea/article/New-COVID-19-variant-increasingly-found-in-15878547.php (2021).
- Wise, J. Covid-19: New coronavirus variant is identified in UK. BMJ 371, m4857 (2020).
- COVID-19 Genomics UK (COG-UK) [email protected]. An integrated national scale SARS-CoV-2 genomic surveillance network. Lancet Microbe 1, e99–e100 (2020).