Introduction
The development of a methodology to assign names to lineages of SARS-CoV-2 using a dynamic nomenclature has previously been described in this bioRxiv article and post on virological.org.
Pangolin (Phylogenetic Assignment of Named Global Outbreak LINeages), the software used to assign lineages, is open source and can be found on github. It was developed with the motivation to make it as easy as possible for labs to obtain useful information from genome sequencing of SARS-CoV-2. For those familiar with the UNIX command line the installation of this software is straightforward requiring just a python interpreter to run pangolin and the conda package management system to install dependencies.
However for those who are unfamiliar with the command line or do not have access to a UNIX computer, a web based application (open source code on gitlab) has been developed to allow users to:
- Assign lineages to genome sequences of SARS-CoV-2
- View descriptive characteristics of the assigned lineage(s)
- View the placement of the lineage in a phylogeny of global samples
- View the temporal and geographic distribution of the assigned lineage(s)
Using the pangolin web application
The URL for the web application is https://pangolin.cog-uk.io
Figure 1 | Initial data loading screen for the pangolin web application
Via this interface, users can upload SARS-CoV-2 genome sequences in single or multi-fasta format via drag and drop or selecting from the browser file browser. After upload, clicking on the ‘Start Analysis’ button the sequences will be analysed with the pangolin software (the versions of pangolin and the underlying data are displayed in the page footer).
Once analysis completes, the lineage for each sample is displayed along with bootstrap and SH-aLRT (Shimodaira–Hasegawa approximate likelihood ratio test) support values. Extra information for that lineage can be displayed including the countries where the lineage is most commonly found, the number of taxa in the global data set matching the lineage, the date range between which the lineage has been observed, and the number of days since the lineage was last sampled.

Figure 2 | Screenshot of the data table post analysis which contains lineage assignment and extra information concerning the lineage. Here one row is shown representing one sample from a multi fasta file.
To help contextualise user samples within the global context, two links to visualisation of the lineage within Microreact (https://www.microreact.org) are shown. One shows where and when sequenced samples of the same lineage have been observed. The second shows the global data filtered for samples from the UK.
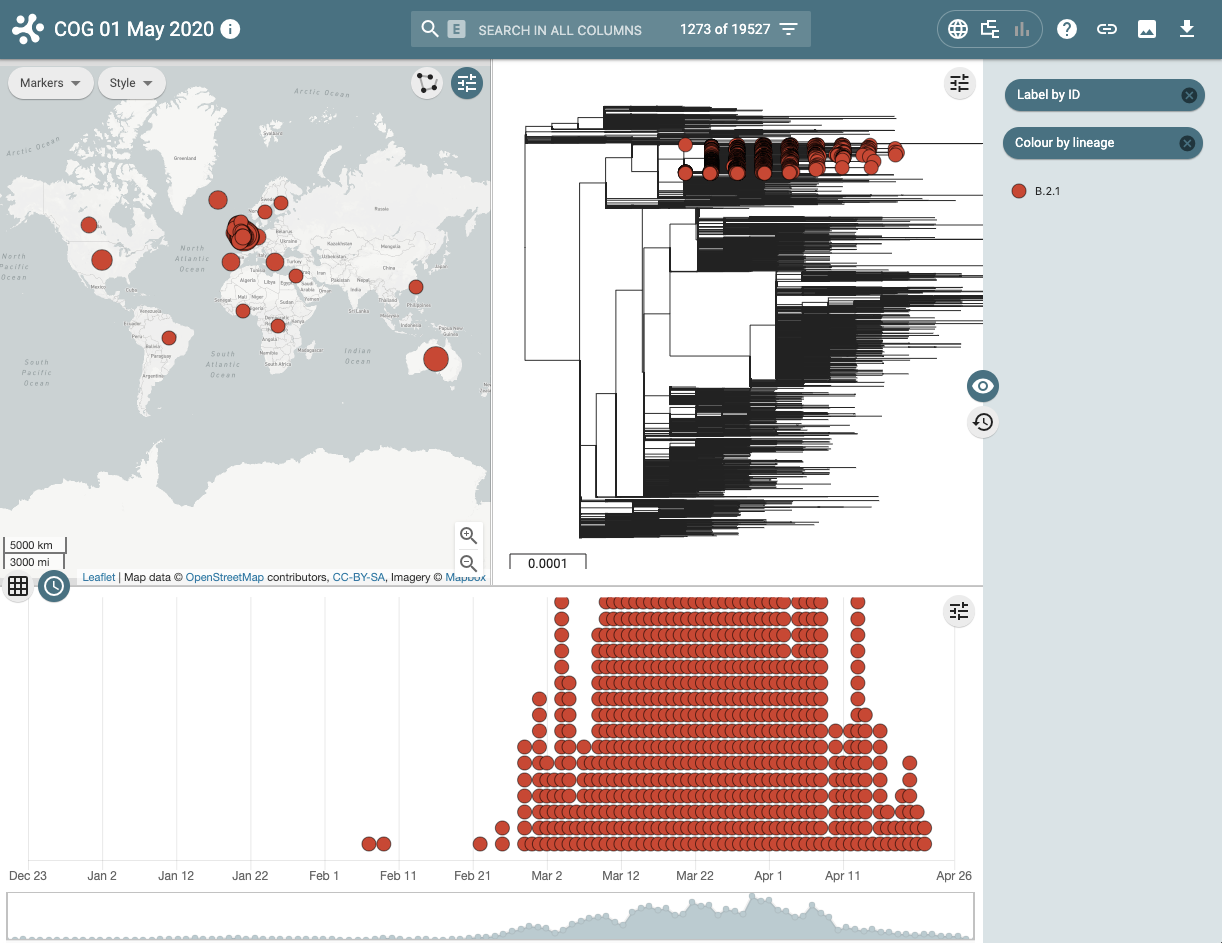
Figure 3 | Microreact visualisation of lineage B.2.1 demonstrating where and when samples have been collected and sequenced.
This visualisation examines the geographical and temporal dynamics of different lineages. For example in the screenshot below it can be seen that lineage A, that contains the earliest ancestors of the SARS-CoV-2 in the human population, although widely distributed, is now less frequently observed in the population since the virus has evolved and the A lineage has been replaced by descendent sub-lineages.
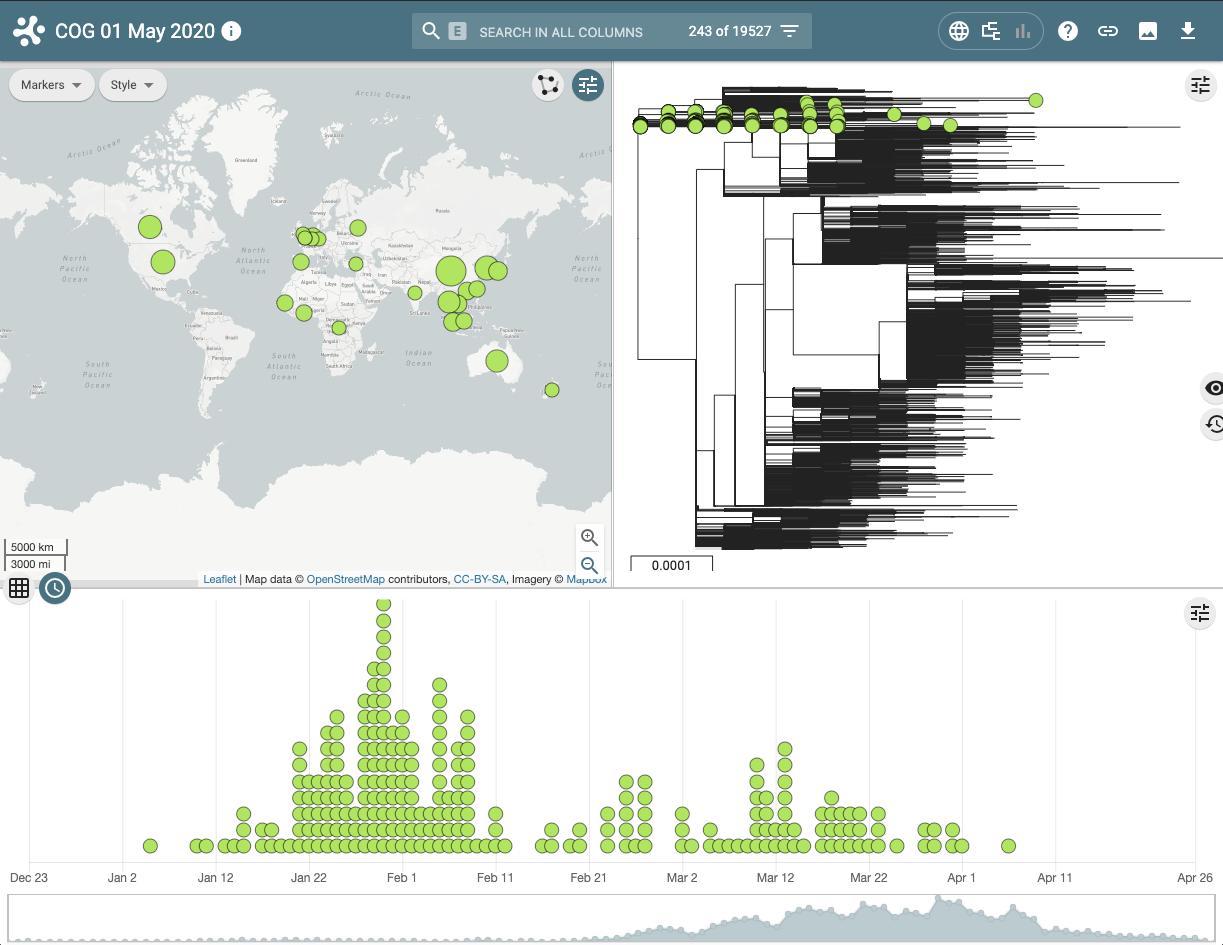
Figure 4 | Microreact visualisation of lineage A demonstrating where and when samples have been collected and sequenced.
Methods
pangolin estimates the most likely placement of a query sequence within a phylogenetic tree of representative sequences, hosted at hCoV-2019/lineages. Internally, pangolin runs mafft (Katoh 2013) and iqtree (Nguyen et al 2015), using the guide tree and alignment to keep analysis overhead relatively lightweight.
The linked Microreact views are informed by publically available genomes from the COG-UK consortium and those submitted to GISAID. We gratefully acknowledge all of the data contributors to GISAID, a list of whom can be found here.
Data privacy
Sequences that are uploaded are discarded once analysis has completed. A hash of the sequence is kept to allow faster processing of identical sequences, but the sequence itself is not stored.
Summary
In summary, the pangolin web application allows assignment of lineages to genome sequences without the end-user requiring a unix computing environment or knowledge of the command line. The results can be downloaded as a CSV spreadsheet file and the lineage assignments contextualised geographically and temporally.
Full instructions on how to use the application can be found https://pangolin.docs.cog-uk.io
For further documentation on how to use Microreact in general see this introductory video and instructions. For an overview and some COVID-19 specific examples see the appendix below
Contributors
Pangolin was developed by Áine O’Toole, JT McCrone, Verity Hill and Andrew Rambaut, Institute of Evolutionary Biology, University of Edinburgh.
The Web application was developed by Khali Abu-Dahab, Ben Taylor, Anthony Underwood, Corin Yeats and David Aanensen from the The Centre for Genomic Pathogen Surveillance team.
The COVID-19 Genomics UK Consortium
References
A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology Andrew Rambaut, Edward C. Holmes, Verity Hill, Áine O’Toole, JT McCrone, Chris Ruis, Louis du Plessis, Oliver G. Pybus. bioRxiv
https://doi.org/10.1101/2020.04.17.046086
Microreact: visualizing and sharing data for genomic epidemiology and phylogeography
Silvia Argimón,Khalil Abudahab, Richard J. E. Goater, Artemij Fedosejev, Jyothish Bhai, Corinna Glasner, Edward J. Feil, Matthew T. G. Holden, Corin A. Yeats, Hajo Grundmann, Brian G. Spratt, and David M. Aanensen. Microbial Genomics 2(11)
https://doi.org/10.1099/mgen.0.000093
A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. L.-T. Nguyen, H.A. Schmidt, A. von Haeseler, B.Q. Minh (2015) IQ-TREE: Mol. Biol. Evol., 32:268-274. https://doi.org/10.1093/molbev/msu300
MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability
Kazutaka Katoh, Daron M. Standley. Molecular Biology and Evolution,30(4):772–780, https://doi.org/10.1093/molbev/mst010
Appendix
Global SARS-CoV-2 Lineage Distribution Visualisation with Microreact
Microreact consists of a number of linked data panels providing an interactive view of underlying data. Within the COG-UK project, global SARS-CoV-2 lineage distributions (as defined by Pangolin), minimal metadata associated with genomes and a global tree indicating genome similarity are available. The map panel indicates location (and frequency) of lineages and the timeline panel, the date associated with each genome record. The tree panel can be switched with a chart panel (from the top right menu) which indicates the distribution of lineages over time (figures 5 and 6) for global data.
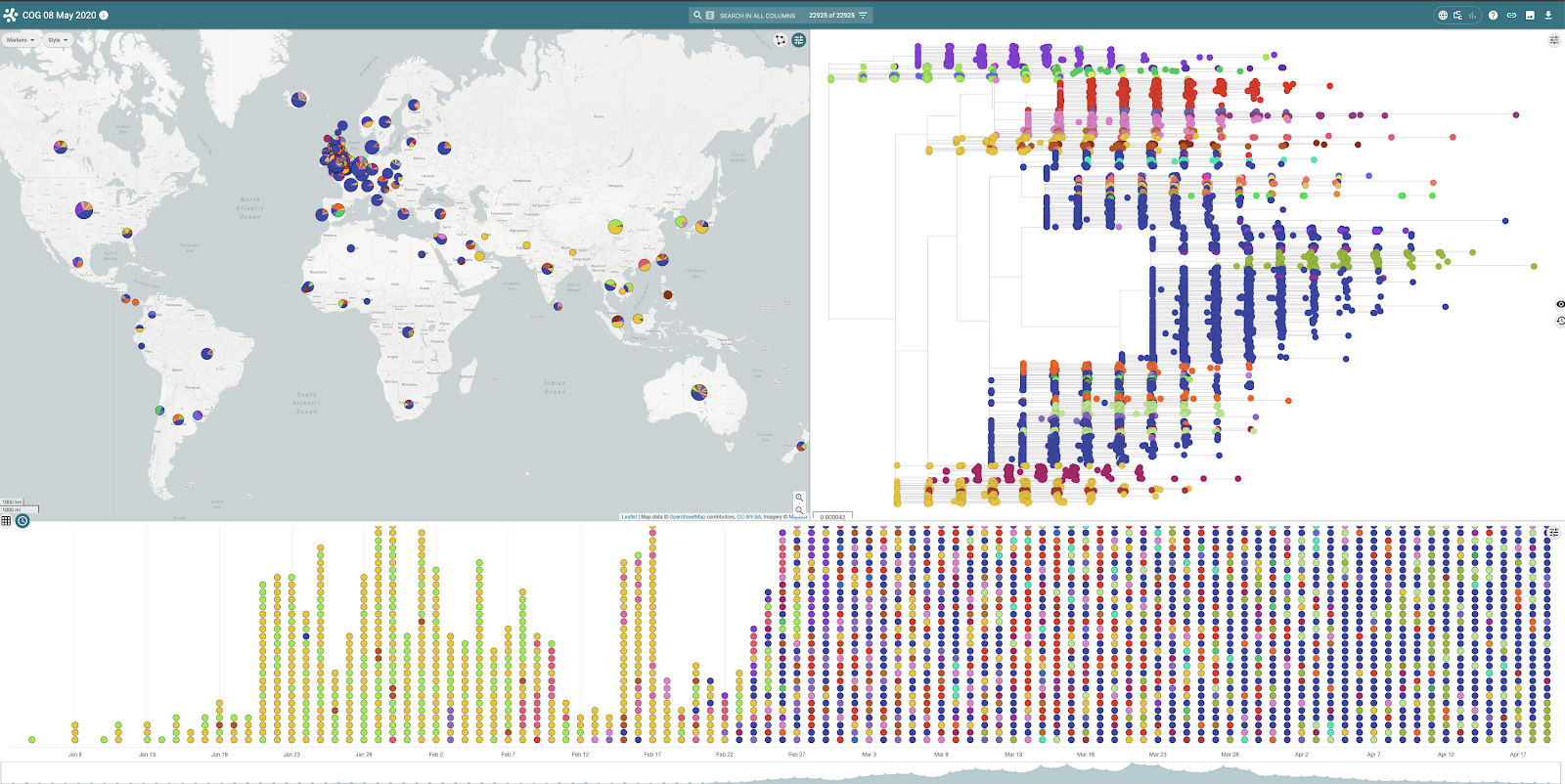
Figure 5 | Global distribution of SARS-Cov2 lineages viewed within Microreact shown on map, tree and timeline

Figure 6 | The tree panel can be switched with a chart panel highlighting lineages and their distribution over time.
Each of the panels can be used to highlight and filter data of interest, for example, selecting a single point on the map highlights the samples from that location on the phylogenetic tree and timeline views.
Map Panel
Selecting a single point on the map highlights samples in other panels from just that location. Additionally, by clicking the map menu (top right of map view) the lasso tool allows a user to click and draw a bounding area around locations of interest which will filter the tree, timeline and chart panels to only show data from the region selected.
Further options are available from the map menu including options for marker style and choice of background tiles - web mapping servers can be linked to to display different tile layers.
Tree Panel
The phylogenetic tree can be zoomed in and out to enable subsets of data to be viewed in detail. Selecting a leaf on the tree will highlight the geographic and temporal location of that genome and selecting an internal node on the tree filters the map, timeline and chart panels to only show data related to the branch selected. Clicking outside of the tree will deselect the filter. Right clicking an internal node displays options for viewing a subtree / rerooting or rotating the branch. Further options are available from the tree menu located at the top right of the tree panel including tree shape and options for nodes and labels.
Chart Panel
The default view shows the distribution of lineages within the dataset and clicking a particular lineage will highlight on the map and timeline all those locations and dates corresponding to the selected lineage. The chart menu, located at the top right of the chart view, indicates the parameters charted allowing a user to change the graph type and axis attributes and explore data further.
Timeline Panel
The timeline shows the temporal position of all data ranging from the first to last dates as points on the timeline.A summary overview is shown below the detailed timeline. Selecting a single point highlights the location of the selected sample within the map and tree panels. The timeline menu, located at the top right of the timeline enables control over the point view and also a set of controls enabling an animated view of data to be ‘played’ with control over the animation speed and time window. Furthermore, the bottom summary view can be used to position the first and last time point displayed in order to filter the samples displayed in other panels.
Data table Panel
The data table can be accessed by clicking the table icon at the top left of the timeline panel. Any column can be filtered by entering text criteria from the filter icon within each column heading, allowing, for example, filtering lineage by a lineage ID. Filtering updates all other data panels.
Saving ‘views’
At any point within exploration of data, the ‘state’ - ie the zoom level of map/tree/timeline along with any filtering of data (via visual or textual filtering) can be saved using the ‘link’ button at the top right of the microreact window, creating a permanent URL which can be shared to allow revisiting of the view without needing to reapply filtering.
Continental Lineage Distribution
By filtering data to show genomes sampled from across Europe eg by using the map ‘lasso’ tool to draw around the continent, the chart panel is updated along with the map and timeline to show, over time, lineages and their distribution (figures 7 and 8)
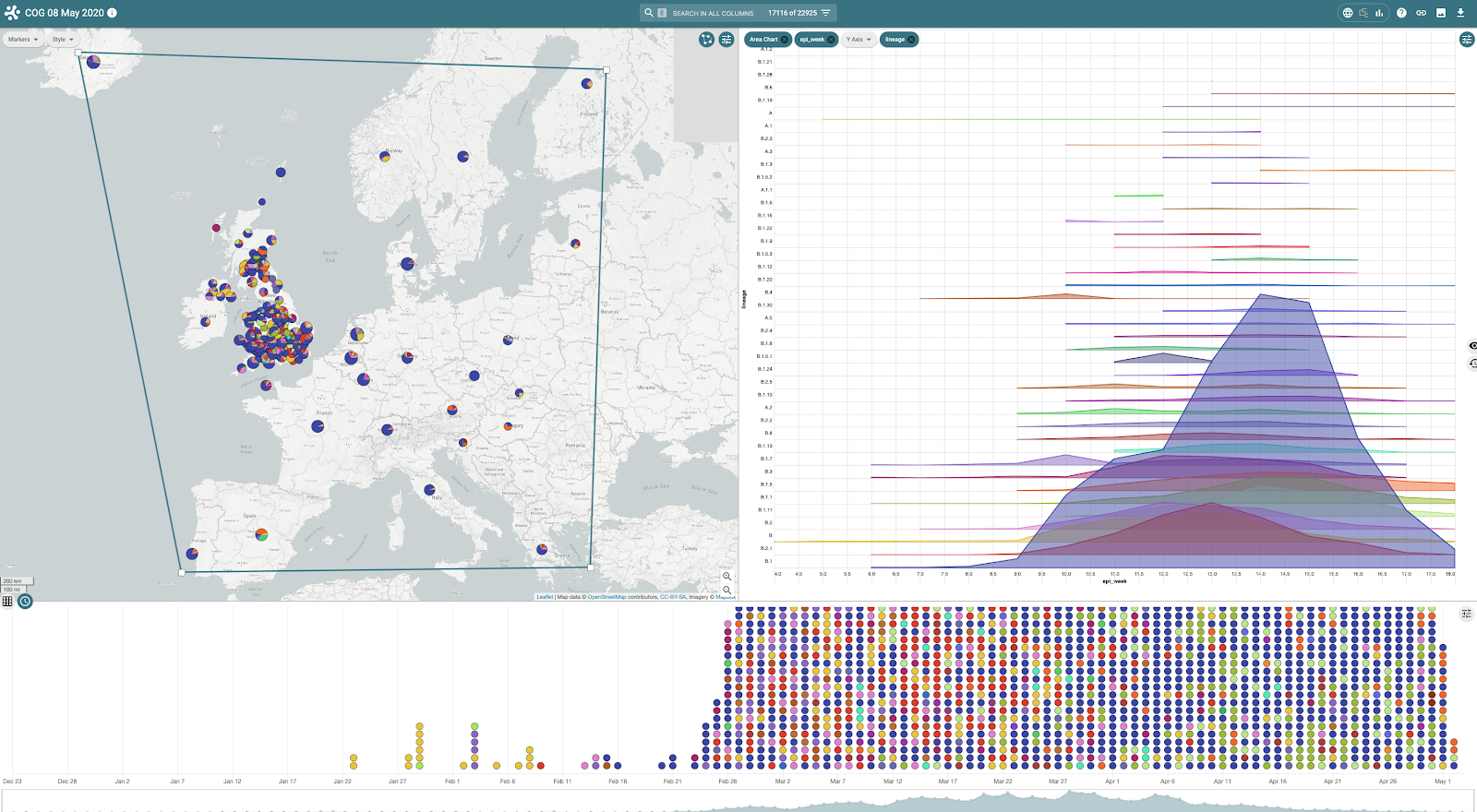
Figure 7 | The chart panel indicates the circulating lineages (Y-axis) over time (epi weeks - X axis) within Europe with the map indicating location of sampled genomes and their frequency per location (pie charts).

Figure 8 | By switching the panel from chart to tree, the location of European genomes are highlighted on the global tree.
Additional features
Data can also be filtered by entering free text in the search bar at the top of the microreact window. Additional features can be accessed from the icons at the top right including exporting images and data and access to a ‘cheat sheet’ detailing microreact controls, an animated tour of microreact features and an issue reporting feature.
Data Distribution and updates
Within the COG-UK consortium, currently data (including trees and metadata) are currently updated on a weekly basis, however automation will move data updates to close to real time to enable the monitoring of trends in lineage distribution and movement. Within the COG-UK consortium, the monitoring of geographic lineage diversity and the consequence of changes in intervention measures and will be enhanced