September 17th 2020
Vegard Eldholm & Ola B Brynildsrud
Norwegian Institute of Public Health
Proponents of the “lab-release theory” for SARS-CoV-2 (SC2) have recently focused their attention on the published bat coronavirus RaTG13 genome, identified in a horseshoe bat (Rhinolophus affinis). The “Yan-report”, which has garnered significant attention recently, claims that the RaTG13 genome is “fake” and generated as a cover-up to prop up the “natural origin theory”. To us, it is far from clear that the existence of RaTG13 has a major bearing on deciding whether SC2 is the result of natural evolution or was created in a laboratory. However, as the genome now seems to play an outsized role in the controversy, it could definitely be harmful to the public’s perception of science if the published genome was found to be some sort of a fabrication.
In their report, Yan et al simply postulate that the published genome is not real, and cite a number of preprints in support of this view. Among the cited preprints, Singla et al. seem to represent a wholehearted attempt to look into the veracity of the published RaTG13 genome. The authors conclude that the RaTG13 genome could not be assembled from the raw sequence data linked to its original publication .
We therefore decided to see for ourselves whether we could recover the RaTG13 genome from the public raw sequence data. Zhou et al. only cursorily describe the methods used for genome assembly: Short metagenomic reads were assembled de novo after depletion of reads of human or animal origin, and gaps in the assembly were filled with Sanger-sequenced amplicons.
We downloaded the reads from ENA (accessions SRR11085797 for the metagenomic data and SRR11806578 for the amplicon data), and separately mapped both short reads and Sanger amplicons to the RaTG13 genome using BWA with default settings. We first inspected the trace in Artemis for poorly covered regions. Singla et al. claim that more than 1% of the genome had zero coverage, but we could not identify any position with a coverage of less than 1. It is possible that Singla et al. refers to the metagenomic sequence only, as SRR11806578 does not seem to be mentioned in the paper. It is essential to keep in mind that Sanger traces are essentially “error-free” when inspected and trimmed properly, which is why the method is still in use for validation of next generation sequencing projects.
It is indeed true that the sequencing depth across RaTG13 is quite low - Our average indicates 9.73, which is exactly the same as Singla’s estimate. This is strange though, since our number was attained WITH the additional coverage from Sanger sequencing. Low depth is to be expected from metagenomic samples, and the Sanger amplicons fill the gaps well. Specifically, the region 13182-13293 claimed by Singla et al to be absent, is indeed covered by a Sanger amplicon closing the gap in the raw read data (Fig. 1).
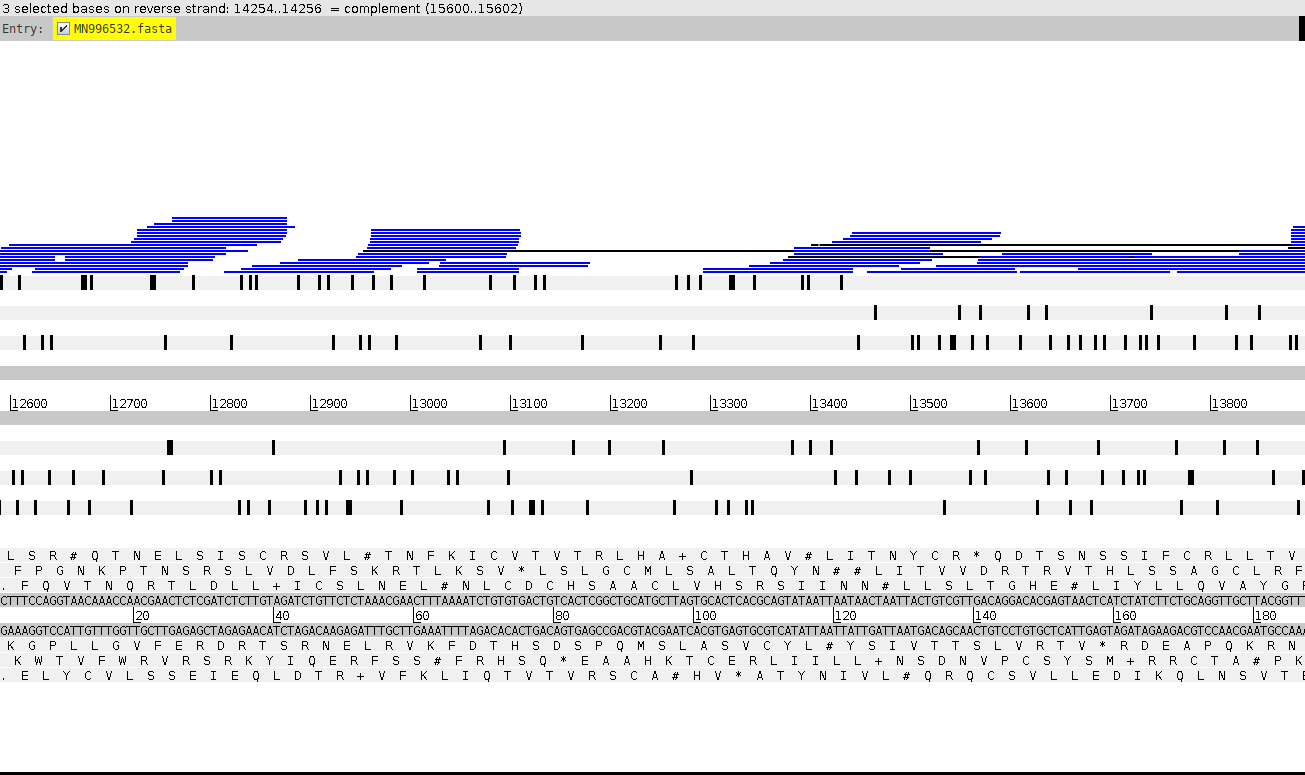
Figure 1. Metagenomic short reads and Sanger amplicons mapped to the RaTG13 genome. Expanded view of a region singled out by Singla et al, demonstrating coverage of assembly gap with Sanger amplicons.
A total of 466 nucleotides in the published genome was not covered by short metagenomic reads in our mapping. On the other hand, a whopping 16540 nucleotides were covered by the Sanger amplicons, illustrating the length to which the authors have gone to patch regions with low/zero depth.
Finally, we used BCFtools to identify potential SNPs between MN996532 and the raw reads generated in Zhou et al’s paper, potentially indicating issues with the data. Across the entire span of the RaTG13 genome, we could only find 2 sites which had basecalls in conflict with the published RaTG13 genome - Positions 18797-18798, which were called as TG rather than GC in our pileups.
To conclude, the published RaTG13 genome is supported by raw sequence data of good quality. It should be noted that our depth estimate of 9.73x above is somewhat misleading, as it counts the essentially error-free Sanger amplicons as “common” error-prone short reads. With Sanger sequencing, a single trace of good quality is generally regarded as sufficient for accurate sequence characterization. The total quality of the data is thus significantly better than the 9.73x depth estimate suggests.