Mutations arising in SARS-CoV-2 spike on sustained human-to-human transmission and human-to-animal passage
Robert F. Garry1,2
1Department of Microbiology and Immunology, Tulane University Medical Center, 1430 Tulane Avenue, New Orleans, Louisiana 70112 USA; E-Mail: [email protected]
2Zalgen Labs, LLC, Germantown, MD, USA
Introduction
The proximal origins of SARS-CoV and MERS-CoV from civets and camels, respectively, are well documented. Few genetic changes in these viruses are required for the interspecies transfers to humans (Li, 2008). While precise details and timing of the evolutionary pathways remain to be elucidated, it is also apparent that SARS-CoV-2 emerged from the Sarbecovirus subgenus of the Betacoronaviruses via one or more interspecies transfers (Andersen et al., 2020; Boni et al., 2020). In contrast to SARS-CoV and MERS-CoV, SARS-CoV-2 has had an extended period of human-to-human transmission. While the evolutionary rate is not unusual for an RNA virus, mutations have occurred that appear to impact SARS-CoV-2 fitness (Kemp et al., 2020; Volz et al., 2020). For example, SARS-CoV-2 carrying G614 has replaced D614 as the predominant circulating variant (Volz et al., 2020). The D614G substitution abolishes a hydrogen-bond interaction with T859 of a neighboring monomer, which destabilizes the spike trimer and increases interaction of the receptor binding domain (RBD) with angiotension-converting enzyme 2ACE2 . By increasing viral load in the upper respiratory tract of COVID-19 patients, D614G may enhance SARS-CoV-2 transmission (Plante et al., 2020).
Recently, a SAR-CoV-2 variant emerged in the UK that has acquired 17 mutations, including 8 in spike (Rambaut et al., 2020). An apparently independent lineage emerged in South Africa that also has multiple spike mutations (Pond et al., 2020). Spike mutations have also occurred during interspecies transfers of SARS-CoV-2 from humans to animals, both during establishment of experimental models of COVID-19 and as an unintended consequence of human interactions with domestic, curated and commercial animals (Mahdy et al., 2020). Here, some of the mutations that have occurred to date in the SARS-CoV-2 spike during human-to-human transmission and following human-to-animal passage are compiled. This compilation highlights several commonly occurring natural features of coronavirus spike evolution that may be involved in interspecies transfers.
Methods
Homology modeling of the SARS-CoV-2 spike was performed in SWISS-MODEL (Waterhouse et al., 2018) using reference sequence QHD43416.1 and a closed prefusion configuration of the spike trimer pdb 6VXX (Walls et al., 2020) as template. The resulting model includes amino acids that are disordered in the spike cryoelectron microscopy structure and reverts the furin cleavage site and proline mutations used to stabilize the trimer. As in structures of all other CoV spikes, this model lacks most of the C-terminal helix (heptad repeat 2), membrane-proximal external region, transmembrane and intracellular domains.
Spike proteins carrying representative mutations that arise on human-to-human passage and human-to-animal passage included:
hCoV-19/SouthAfrica/Tygerberg-461/2020|EPI_ISL_745186|2020-12-07
hCoV-19/England/LOND-1267020/2020|EPI_ISL_741243|2020-12-11
hCoV-19/mouse/Harbin/HRB-26m/2020|EPI_ISL_459910|2020-04-19
hCoV-19/mink/Netherlands/1/2020|EPI_ISL_431778|2020-04-24
hCoV-19/mink/Netherlands/NB01_02KS/2020|EPI_ISL_447624|2020-04-29
hCoV-19/mink/Netherlands/NB02_07KS/2020|EPI_ISL_447629|2020-04-29
hCoV-19/mink/Netherlands/NB02_16RS/2020|EPI_ISL_447632|2020-04-28
hCoV-19/cat/France/Env-Ba/2020|EPI_ISL_483063|2020-05-14
hCoV-19/cat/France/Env-Di/2020|EPI_ISL_483064|2020-05-14
hCoV-19/cat/Belgium/BE-MG-0320/2020|EPI_ISL_487275|2020-03-11
hCoV-19/cat/Denmark/mDK-315/2020|EPI_ISL_683164|2020-11-17
hCoV-19/cat/USA/TX-TAMU-013/2020|EPI_ISL_699506|2020-06-28
hCoV-19/cat/USA/TX-TAMU-057/2020|EPI_ISL_699507|2020-07-17
hCoV-19/cat/USA/TX-TAMU-078/2020|EPI_ISL_699509|2020-07-29
hCoV-19/cat/Greece/2K/2020|EPI_ISL_717979|2020-11-23
hCoV-19/lion/USA/NY-3-041520/2020|EPI_ISL_566037|2020-04-04
hCoV-19/lion/USA/NY-041520/2020|EPI_ISL_566038|2020-04-04
hCoV-19/lion/USA/NY-2/2020|EPI_ISL_566044|2020-04-04
hCoV-19/tiger/USA/NY-040420/2020|EPI_ISL_420293|2020-04-02
hCoV-19/dog/HongKong/20-02756/2020|EPI_ISL_414518|2020-02-26
hCoV-19/dog/USA/TX-TAMU-077/2020|EPI_ISL_699508|2020-07-28
hCoV-19/dog/Italy/Dog399-20BA/2020|EPI_ISL_730652|2020-11-04,
Spike amino acid sequences were aligned using Clustal Omega (Sievers et al., 2011).
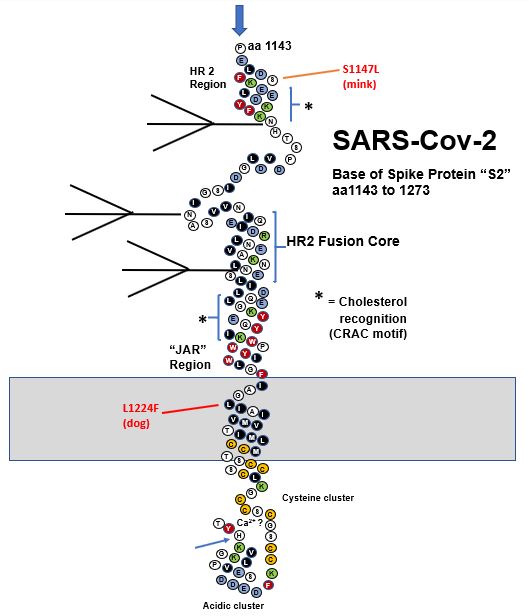
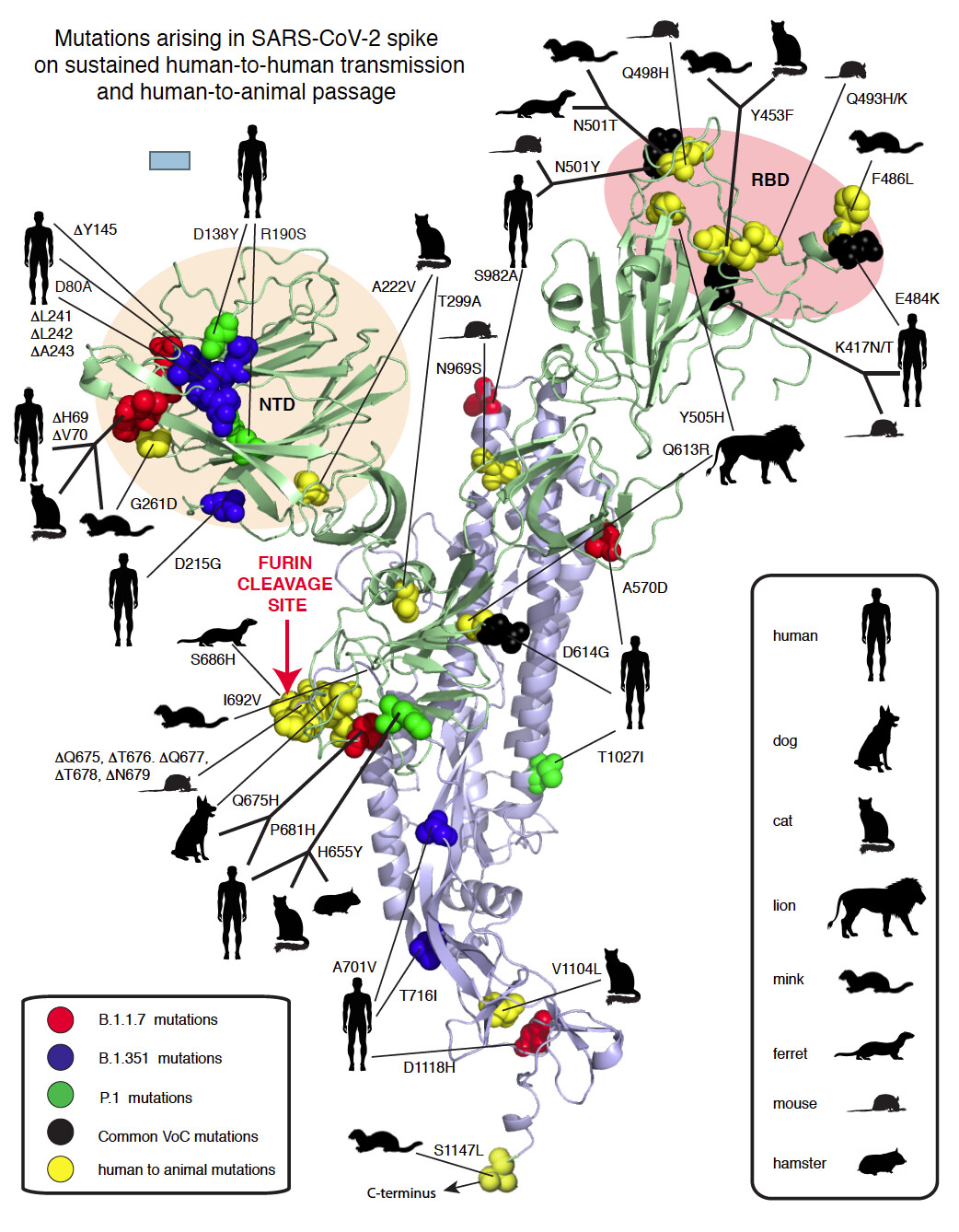
Figure 1. Compilation of SARS-CoV-2 spike mutations occurring in humas and animals. Red spheres: United Kingdom (UK) variant, Blue spheres: South African (ZA) variant, Magenta: both UK/ZA variants, Yellow spheres: animals as indicated in the inset. NTD: Amino-terminal domain. RBD: Receptor binding domain.
Results
Mutation of two amino acids (K479N and S487T) in palm civet SARS-CoV RBD allows this virus to infect humans (Li, 2008). These changes overcome the species barrier between civets and humans and enable favorable interactions between the RBD and contact residues on human ACE2. Q493 and N501 are the two SARS-CoV-2 residues that have similar interactions with ACE2 as SARS-CoV residues N479 and T487. The UK variant (Variant of Concern 202012/01, B.1.1.7) contains spike mutations N501Y, as well as delH69, delV70, delY145, A570D, P681H, T716I, S982A and D1118H and is estimated to have emerged in the UK during the Fall of 2020 (Fig. 1). N501Y likely enables a Pi-Pi interaction between Spike 501Y and ACE2 41Y. The South African (ZA) variant (501Y.V2, B.1.351) also includes N501Y plus variously D80A, D215G, K417N, E484K and A701V (Fig. 1). The UK and ZA variants have now been detected elsewhere around the world. The African Centre of Excellence for Genomics of Infectious Diseases (ACEGID), Redeemer’s University, Nigeria, identified two examples of an SARS-CoV-2 spike that shares the P681H in common with the UK variant. This mutation alters the out-of-frame insertion (relative to other sarbecoviruses) that generates the minimal furin cleavage site in SARS-CoV-2 spike (Happi et al., 2020).
A variety of interspecies transfers of SARS-CoV-2 from humans to animals have occurred. (Fig. 1) The N501Y mutation, observed in the UK and ZA variants was selected in 6 passages in aged mice and enables efficient replication of SARS-CoV-2 (Sun et al., 2020). Further passages of the N501Y mutant resulted in selection of Q493H and K417N, which increased pathogenicity the mouse model. K417N is also observed in the ZA variant. Q493K, a related mutation to Q493H, also increases both replication and pathogenesis in mice (Leist et al., 2020). Another mouse strain (EPI_ISL_459910) generated by six passages is also characterized by Q493K. This variant also carries a deletion of amino acids Q675 to N679 that appears to have occurred after a single passage in Vero cells (Liu et al., 2020).
Humans with SARS-CoV-2 have infected minks on fur farms in several countries (Koopmans, 2021). Recurrent mutations observed in mink include Y453F, F486L and N501T, which are all in the RBD (Lassaunière et al., 2020; Oude Munnink et al., 2020) (Fig. 1). As is the case with N501Y in humans and mice, the N501T change may enhance binding to mink ACE2. The same N501T mutation was observed in passage of SARS-Cov2 ferret, another mustelid (Richard et al., 2020). Two furin cleavage site mutations have occurred in mink and ferrets, I692 V and S686 respectively.
Several examples of transfer to domestic cats have been documented (Braun et al., 2020; Hamer et al., 2020; Neira et al., 2020; Wu et al., 2020). Felids in zoos have also been infected with SARS-CoV-2, presumably via contact with humans. Dogs are also permissive for infection by SARS-CoV-2 (Hamer et al., 2020; Sit et al., 2020). A variety of mutations in SARS-CoV-2 after interspecies transfers to felines or canines have been observed some of which are common to other interspecies transfers (Fig. 1). However, none of the mutations appear essential for replication in these species. A sequence from a zoo tiger did not show any mutations (Wang et al., 2020).
Discussion
The mutations compiled above fall into four general classes:
- RBD mutations, which are of importance because some may provide both immune escape and a fitness advantage. As previously documented for SARS-CoV (Li, 2008), RBD mutations, as exemplified by N501Y/T are also important for interspecies transfer.
- The amino terminal domain (NTD), particularly the portion most exposed on the virion surface, represents another hotspot for spike mutations. There is evidence for immune selection in this region and preliminary evidence that at least one of these changes delH69/delV70 could improve fitness (Kemp et al., 2020).
- Variation in or near the FCS is a common natural occurrence in CoV evolution (Gallaher and Garry, 2020). The changes during transfer of SARS-CoV-2 to species provide additional examples of the importance of the spike cleavage sites at S1/S2 junction and S2’ interspecies transfer. The impact of specific changes such as a P681H on transmissibility are important to determine.
- Several spike mutations group with D614G, including Q613R found in lions. Mutations that occur in the metastable regions of spike could influence refolding to the 6-helix bundle during virus entry possibly affecting infection efficiency. Another mutation cluster in a metastable region is at the base of the structural model that involves the prefusion to postfusion transition of spike and is also of importance for neutralizing antibodies in other class I viral fusion proteins (Hastie et al., 2017).
Conclusions
Elucidation of the mechanisms by which viruses adapt to different hosts thereby crossing species barriers is important for identifying potential epizootic threats. Widespread transmission of an emerging pathogen, such as SARS-Cov-2 can potentially lead to further mutations that affect transmissibility or effectiveness of countermeasures. Infrastructure for continuous monitoring of infectious viral diseases, as implemented in the UK, should be enabled worldwide to response to such changes. Monoclonal antibody immunotherapeutics should be formulated as cocktails to prevent mutational escape to single antibodies (Baum et al., 2020). Vaccines should be designed to maximize polyclonal immune responses to multiple protective epitopes.
Acknowledgments
I am grateful to scientists worldwide who have made SARS-CoV-2 genome sequences available to the research community prior to publication. Kristian G. Andersen, Edward C. Holmes and Andrew Rambaut provided essential input and discussion. Work on emerging viruses in the Garry Laboratory is supported by the National Institutes of Health, the Coalition for Epidemic Preparedness Innovations, the Burroughs Wellcome Fund, the Wellcome Trust, the Center for Disease Prevention and Control, and the European & Developing Countries Clinical Trials Partnership.
References
Andersen, K.G., Rambaut, A., Lipkin, W.I., Holmes, E.C., and Garry, R.F. (2020). The proximal origin of SARS-CoV-2. Nat Med 26, 450-452.
Baum, A., Fulton, B.O., Wloga, E., Copin, R., Pascal, K.E., Russo, V., Giordano, S., Lanza, K., Negron, N., Ni, M., et al. (2020). Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 369, 1014-1018.
Boni, M.F., Lemey, P., Jiang, X., Lam, T.T., Perry, B.W., Castoe, T.A., Rambaut, A., and Robertson, D.L. (2020). Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nature microbiology 5, 1408-1417.
Braun, K.M., Moreno, G.K., Halfmann, P.J., Baker, D.A., Boehm, E.C., Weiler, A.M., Haj, A.K., Hatta, M., Chiba, S., Maemura, T., et al. (2020). Transmission of SARS-CoV-2 in domestic cats imposes a narrow bottleneck. Preprint. https://europepmc.org/article/MED/33236011
Gallaher, W.R., and Garry, R.F. (2020). Naturally occurring indels in multiple coronavirus spikes. https://pando.tools/t/naturally-occurring-indels-in-multiple-coronavirus-spikes/560.
Hamer, S.A., Pauvolid-Corrêa, A., Zecca, I.B., Davila, E., Auckland, L.D., Roundy, C.M., Tang, W., Torchetti, M., Killian, M.L., Jenkins-Moore, M., et al. (2020). Natural SARS-CoV-2 infections, including virus isolation, among serially tested cats and dogs in households with confirmed human COVID-19 cases in Texas, USA. bioRxiv : https://www.biorxiv.org/content/10.1101/2020.12.08.416339v1.
Happi, C., Ihekweazu, C., Nkengasong, J., Oluniyi, P.E., and Olawoye, I. (2020). Detection of SARS-CoV-2 P681H Spike Protein Variant in Nigeria. https://pando.tools/t/detection-of-sars-cov-2-p681h-spike-protein-variant-in-nigeria/567.
Hastie, K.M., Zandonatti, M.A., Kleinfelter, L.M., Heinrich, M.L., Rowland, M.M., Chandran, K., Branco, L.M., Robinson, J.E., Garry, R.F., and Saphire, E.O. (2017). Structural basis for antibody-mediated neutralization of Lassa virus. Science 356, 923-928.
Kemp, S.A., Harvey, W.T., Datir, R.P., Collier, D.A., Ferreira, I., Carabelli, A.M., Robertson, D.L., and Gupta, R.K. (2020). Recurrent emergence and transmission of a SARS-CoV-2 Spike deletion ΔH69/V70. bioRxiv 2020.12.14.422555; doi: https://doi.org/10.1101/2020.12.14.422555
Koopmans, M. (2021). SARS-CoV-2 and the human-animal interface: outbreaks on mink farms. The Lancet Infectious diseases 21, 18-19.
Lassaunière, R., Fonager, J., Rasmussen, M., Frische, A., Strandh, C.P., Rasmussen, T.B., Bøtner, A., and Fomsgaard, A. (2020). Working paper on SARS-CoV-2 spike mutations arising in Danish mink, their spread to humans and neutralization data. https://files.ssi.dk/Mink-cluster-5-short-report_AFO2.
Leist, S.R., Dinnon, K.H., 3rd, Schäfer, A., Tse, L.V., Okuda, K., Hou, Y.J., West, A., Edwards, C.E., Sanders, W., Fritch, E.J., et al. (2020). A Mouse-Adapted SARS-CoV-2 Induces Acute Lung Injury and Mortality in Standard Laboratory Mice. Cell 183, 1070-1085.e1012.
Li, F. (2008). Structural analysis of major species barriers between humans and palm civets for severe acute respiratory syndrome coronavirus infections. J Virol 82, 6984-6991.
Liu, Z., Zheng, H., Lin, H., Li, M., Yuan, R., Peng, J., Xiong, Q., Sun, J., Li, B., Wu, J., et al. (2020). Identification of Common Deletions in the Spike Protein of Severe Acute Respiratory Syndrome Coronavirus 2. J Virol 94.:e00790-20. doi: 10.1128/JVI.00790-20.
Mahdy, M.A.A., Younis, W., and Ewaida, Z. (2020). An Overview of SARS-CoV-2 and Animal Infection. Frontiers in veterinary science 7, 596391.
Neira, V., Brito, B., Agüero, B., Berrios, F., Valdés, V., Gutierrez, A., Ariyama, N., Espinoza, P., Retamal, P., Holmes, E.C., et al. (2020). A household case evidences shorter shedding of SARS-CoV-2 in naturally infected cats compared to their human owners. Emerging microbes & infections, 1-22.
Oude Munnink, B.B., Sikkema, R.S., Nieuwenhuijse, D.F., Molenaar, R.J., Munger, E., Molenkamp, R., van der Spek, A., Tolsma, P., Rietveld, A., Brouwer, M., et al. (2020). Transmission of SARS-CoV-2 on mink farms between humans and mink and back to humans. Science. epublication 10.1126/science.abe5901.
Plante, J.A., Liu, Y., Liu, J., Xia, H., Johnson, B.A., Lokugamage, K.G., Zhang, X., Muruato, A.E., Zou, J., Fontes-Garfias, C.R., et al. (2020). Spike mutation D614G alters SARS-CoV-2 fitness. Nature. epublication 10.1038/s41586-020-2895-3.
Pond, S.L., K., Wilkison, E., Weaver, S., Jame, S.E., Tegally, H., de Oliveira, T., and Martin, D. (2020). A preliminary selection analysis of the South African V501.V2 SARS-CoV-2 clade. https://pando.tools/t/a-preliminary-selection-analysis-of-the-south-african-v501-v2-sars-cov-2-clade/573.
Rambaut, A., Loman, N., Pybus, O., Barclay, W., Barrett, J., Carabelli, A., Connor, T.R., Peacock, T., Robertson, D.L., and Volz, E. (2020). Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. https://pando.tools/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563/5.
Richard, M., Kok, A., de Meulder, D., Bestebroer, T.M., Lamers, M.M., Okba, N.M.A., Fentener van Vlissingen, M., Rockx, B., Haagmans, B.L., Koopmans, M.P.G., et al. (2020). SARS-CoV-2 is transmitted via contact and via the air between ferrets. Nature communications 11, 3496.
Sievers, F., Wilm, A., Dineen, D., Gibson, T.J., Karplus, K., Li, W., Lopez, R., McWilliam, H., Remmert, M., Söding, J., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular systems biology 7, 539.
Sit, T.H.C., Brackman, C.J., Ip, S.M., Tam, K.W.S., Law, P.Y.T., To, E.M.W., Yu, V.Y.T., Sims, L.D., Tsang, D.N.C., Chu, D.K.W., et al. (2020). Infection of dogs with SARS-CoV-2. Nature 586, 776-778.
Sun, S., Gu, H., Cao, L., Chen, Q., Yang, G., Li, R.-T., Fan, H., Ye, Q., Deng, Y.-Q., Song, X., et al. (2020). Characterization and structural basis of a lethal mouse-adapted SARS-CoV-2. bioRxiv doi: https://doi.org/10.1101/2020.11.10.377333.
Volz, E., Hill, V., McCrone, J.T., Price, A., Jorgensen, D., O’Toole, Á., Southgate, J., Johnson, R., Jackson, B., Nascimento, F.F., et al. (2020). Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell.
doi: 10.1016/j.cell.2020.11.020. Online ahead of print.
Walls, A.C., Park, Y.J., Tortorici, M.A., Wall, A., McGuire, A.T., and Veesler, D. (2020). Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 181, 281-292.e286.
Wang, L., Mitchell, P.K., Calle, P.P., Bartlett, S.L., McAloose, D., Killian, M.L., Yuan, F., Fang, Y., Goodman, L.B., Fredrickson, R., et al. (2020). Complete Genome Sequence of SARS-CoV-2 in a Tiger from a U.S. Zoological Collection. Microbiol Resour Announc 9. epublication 10.1128/mra.00468-20
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., Heer, F.T., de Beer, T.A.P., Rempfer, C., Bordoli, L., et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46, W296-w303.
Wu, L., Chen, Q., Liu, K., Wang, J., Han, P., Zhang, Y., Hu, Y., Meng, Y., Pan, X., Qiao, C., et al. (2020). Broad host range of SARS-CoV-2 and the molecular basis for SARS-CoV-2 binding to cat ACE2. Cell discovery 6, 68.