Preliminary reconstruction of the current MV Hondius Andes virus outbreak sequences
Note on data use
This is an informal analysis of currently available consensus sequences and is not intended as a formal preprint or manuscript in its current form or with its current authorship. It should not affect sequence generators’ ability to publish with priority any analyses based on the data they generated.
We are in the process of contacting the original data submitters to coordinate any formal preprint or publication arising from this work. We thank all laboratories and researchers who generated and rapidly shared the sequence data that made this analysis possible. We also thank the Pathoplexus team for rapidly enabling Andes virus sequence sharing, and Dr David Safronetz and Dr Jonathan Audet for helpful discussions on the two Canadian sequences.
Summary
We analysed the S, M and L segment consensus sequences from the current MV Hondius-associated Andes virus outbreak available as of 17 May 2026. Our aim here was to reconstruct the relationships among the outbreak-associated sequences, identify candidate differences among them, and assess whether the available consensus data support a likely root of the sampled outbreak diversity.
Our main findings are:
-
As of 19 May 2026, the sampled outbreak diversity is compatible with PP_006WDKH.1 as the root of the current outbreak. Based on consensus-sequence comparisons of the eight currently available outbreak sequences, we identified several candidate differences across the S, M and L segments (Figure 1). All resolved coding-region differences were synonymous, including L:A3437G, L:C2144T, and the additional M and L segment changes observed in PP_006WDKH.2 .
-
Several non-coding or terminal differences should be treated cautiously. Candidate differences including S:G28/29A, M:G51A, and M:C3500T occur near poorly resolved alignment regions and may represent sequencing, assembly or consensus-calling artefacts pending raw-read validation.
-
Most outbreak sequences are nearly identical. With the exception of the divergent
XDHK.2consensus, the currently available outbreak sequences differ by very few candidate mutations. This supports placing the root of the sampled outbreak close to the earliest available sequence, collected on 26 April 2026. -
An ambiguous M segment indel-associated signal could affect the precise rooting. PP_006W6RC.2 carries a T state at M:3500 and a nearby partially resolved insertion pattern also present in the closest non-outbreak comparator sequences. This raises the possibility that
W6RC.2could represent an alternative root or near-root sequence. However, because this signal occurs in a poorly resolved terminal non-coding region, raw-read-level validation is required before revising the rooting.
Update since 17 May 2026: Since this analysis was performed, the Canadian consensus sequences have been updated on Pathoplexus. The previously divergent PP_006XDHK.2 sequence has been replaced by PP_006XDHK.4, which now clusters more closely with the other Canadian sequence, PP_006XDJH.4. However, the two Canadian sequences still differ from one another at multiple sites, including L:1841, L:3965, L:4427, L:4544; M:90, M:651; and S:927, S:930, S:1799, and XDHK.4 remains relatively divergent compared with the other outbreak-associated consensus sequences. Notably, in the updated sequences, XDJH.4 is no longer identical to the French sample, PP_006XBKH.1: both Canadian sequences now carry three additional S segment differences at S:927, S:930, and S:1799.
Data curation and alignment
We downloaded Andes virus sequences for the S, M and L genome segments from ENA, Pathoplexus and GenBank, including eight outbreak-associated sequences available as of 17 May 2026:
PP_006WDKH.1 (WDKH.1), PP_006WDJK.1 (WDJK.1), PP_006W6RC.2 (W6RC.2), PP_006WBLH.1 (WBLH.1), PP_006W3U9.2 (W3U9.2), PP_006XBKH.1 (XBKH.1), PP_006XDHK.2 (XDHK.2), and PP_006XDJH.2 (XDJH.2).
PP_006WANE.2 (WANE.2) was excluded because it was recently revoked from Pathoplexus.
Sequences were aligned to the reference genomes NC_003466.1 for the S segment, NC_003467.2 for the M segment, and NC_003468.2 for the L segment using MAFFT v7.5 with the --keeplength option. We then manually inspected the consensus alignments, focusing on the outbreak-associated sequences because only a small number of candidate differences separate them.
Several positions were masked or treated cautiously before downstream interpretation:
- In the S segment, positions 22, 28, 1823-1824, and the terminal region from 1850 onwards showed patterns consistent with local alignment artefacts, ambiguous bases or poor end-of-sequence resolution.
- In the M segment, position 10 in
WDJK.1and positions 3651-3652 were considered unreliable. - In the L segment, the
Gat position 10 inWDKH.1was treated as a one-base alignment shift and readjusted accordingly.
Outbreak mutation reconstruction
The relationships among the eight available outbreak sequences are compatible with taking WDKH.1|2026-04-26, the earliest available sequence, as the root of the sampled outbreak diversity (Figure 1).
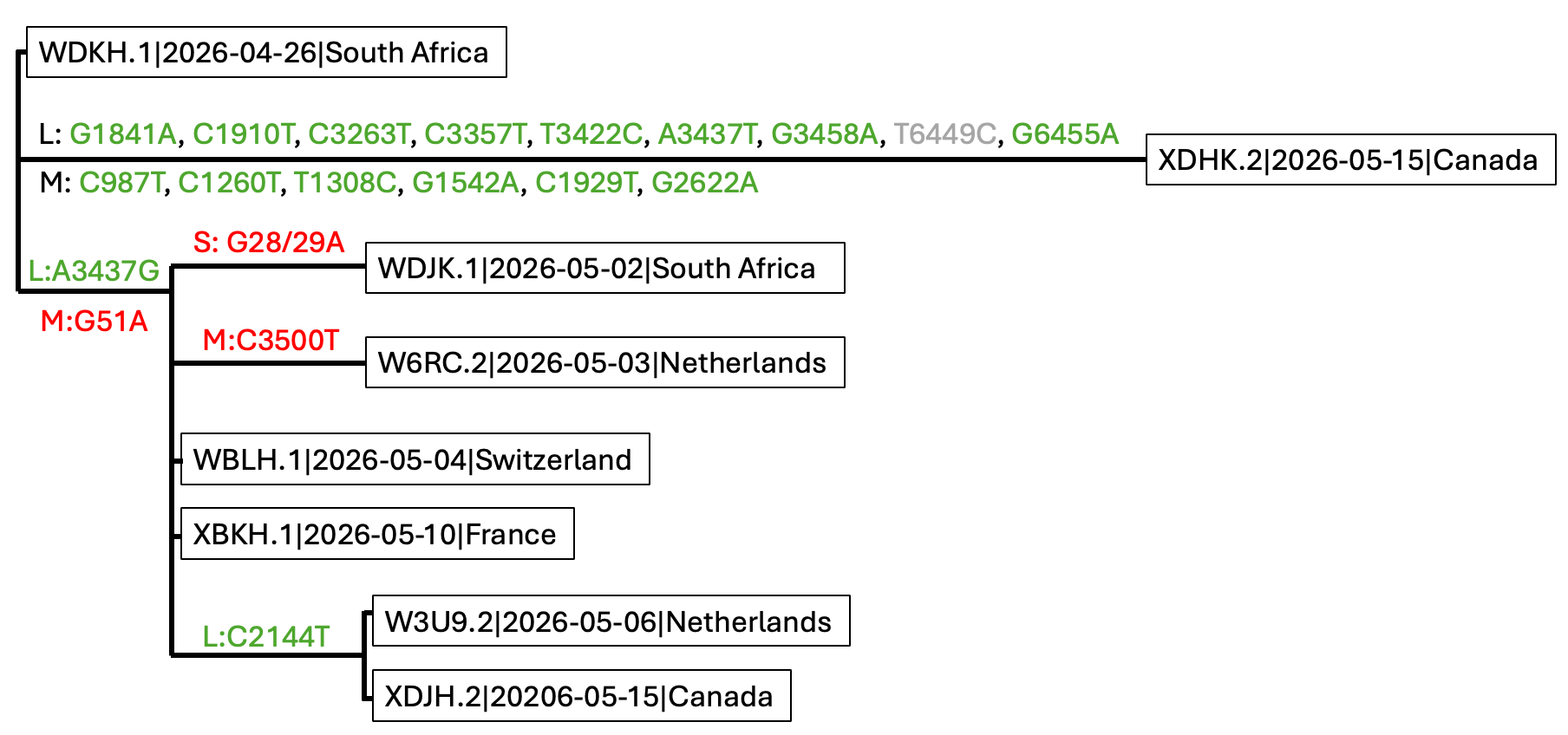
Figure 1. Manually reconstructed mutation tree of the eight available outbreak-associated Andes virus sequences as of 17 May 2026. The topology was constructed from the observed genetic similarity among samples after masking likely alignment or sequencing artefacts, rather than inferred using a phylogenetic dating program. Mutations are annotated with respect to reference genomes NC_003466.1 for the S segment, NC_003467.2 for the M segment, and NC_003468.2 for the L segment. Green mutations are synonymous coding changes. L:T6449C could not be resolved at the amino-acid level because the corresponding codon in
WDKH.1 contains an ambiguous base. Red mutations are non-coding or terminal candidate differences and may represent sequencing or consensus-calling artefacts pending raw-read validation.
After masking likely artefacts, we identified the following candidate differences among the outbreak sequences, excluding the highly divergent XDHK.2 consensus:
M segment: G51A, C3500T
L segment: A3437G, C2144T
The two L segment changes, L:A3437G and L:C2144T, fall within the annotated coding region and are synonymous relative to WDKH.1. The M-segment differences M:G51A and M:C3500T are non-coding or terminal and should be treated cautiously.
We note that a previous Virological post based on raw sequencing reads reported that the S and M segment outbreak sequences were identical and retained only two SNPs in the L segment as true differences among outbreak sequences. Our analysis is based on consensus sequences only and does not include raw-read-level validation. Therefore, the non-coding or terminal M-segment differences should not be interpreted as confirmed SNPs without further validation.
Ambiguous sites affecting outbreak rooting
Several ambiguous sites could affect the rooting of the outbreak sequences and should be interpreted cautiously.
First, the earliest collected sample, WDKH.1, contains a G at M-segment reference position 51, corresponding to the candidate mutation M:G51A in other outbreak sequences. However, this site is immediately followed by a missing base in the WDKH.1 consensus sequence and lies adjacent to the beginning of the M-segment coding region. If interpreted literally, the G state would alter the expected ATG start codon of the M-encoded open reading frame to GTG. We therefore consider this site likely to reflect a sequencing, consensus-calling or local assembly artefact rather than a reliable biological difference.
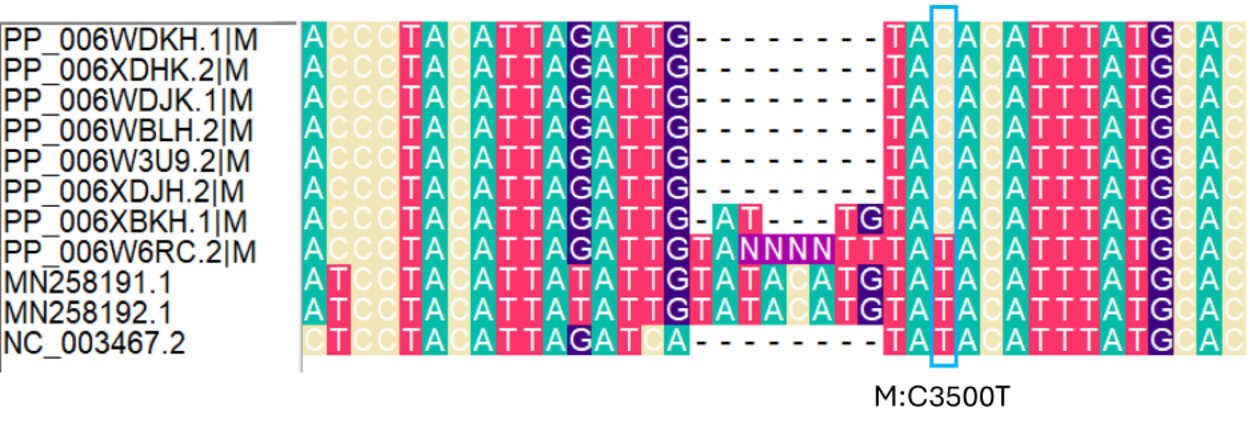
Second, W6RC.2 has a T at M-segment position 3500 (M:C3500T), located in the 3′ non-coding region of the M segment. This T state is also present in the closest non-outbreak comparator sequences and in the reference sequence NC_003467.2 (Figure 2A). Two nucleotides upstream of position 3500, W6RC.2 also contains a partially resolved 8-nucleotide insertion that is absent from the reference genome but present in the closest available non-outbreak sequences. The outbreak sequence XBKH.1 contains a shorter 4-nucleotide insertion at the same coordinates.
Taken at face value, the combination of M:3500T and the upstream insertion in W6RC.2 could suggest an alternative rooting in which W6RC.2 is placed closer to the non-outbreak comparator sequences, discordant with its sampling date. However, because this signal involves an indel in a poorly resolved terminal region, raw-read analysis is required to determine whether the insertion is genuine rather than a sequencing or assembly artefact.
Third, XDHK.2 is unusually divergent, particularly in the L segment, despite presumably deriving from the same Canadian patient as XDJH.2. Most of the additional differences in XDHK.2 are unique to that sequence. However, two L-segment sites overlap with states observed in the closest non-outbreak comparator sequences: L:3263T and L:3437T (Figure 2B). Position 3263 lies immediately upstream of a long stretch of ambiguous bases and is unresolved in XDJH.2. Position 3437 is variable across the alignment, with T in XDHK.2 and the comparator sequences, A in WDKH.1, and G in the remaining outbreak sequences.
Although these patterns raise the possibility that XDHK.2 retains outgroup-like states at some sites, the combination of unique mutations, nearby ambiguity and missing data means that we cannot confidently interpret XDHK.2 as an outgroup to the remaining outbreak sequences.
(A)
(B)
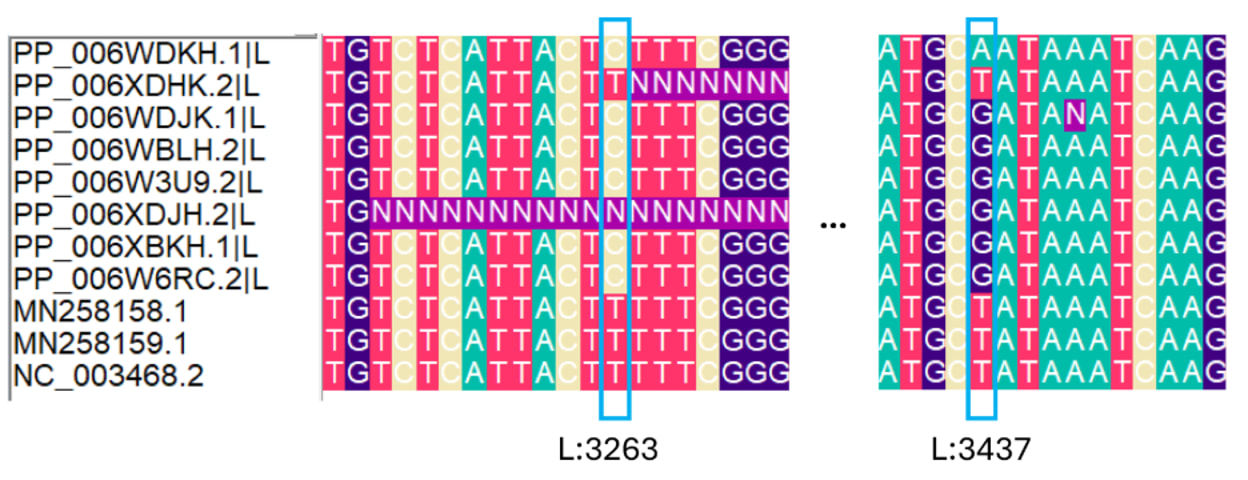
Figure 2. Ambiguous alignment features affecting possible rooting of the outbreak sequences. Nucleotide alignments of selected M (A) and L segment (B) regions showing sites that may influence the inferred rooting of the outbreak cluster. In the M segment,
W6RC.2 carries M:C3500T in the 3′ non-coding region, matching the closest non-outbreak comparator sequences and the reference sequence NC_003467.2, and also shows a partially resolved upstream insertion. XBKH.1 carries a shorter insertion at the same region. In the L segment, XDHK.2 carries states matching comparator sequences at positions 3263 and 3437, but position 3263 is adjacent to a stretch of ambiguous bases and position 3437 is variable across the outbreak alignment. These sites may indicate alternative rooting signals, but they occur in regions affected by ambiguity or indel variation and require raw-read-level validation before being interpreted as genuine phylogenetic signal.
Interpretation
The available outbreak-associated consensus sequences are extremely similar, with the exception of the divergent XDHK.2 consensus. Under the masked consensus alignment, the sampled outbreak root is compatible with the earliest available sequence, WDKH.1, collected on 26 April 2026. We consider this the most parsimonious rooting based on the current consensus data.
However, several candidate differences that could affect rooting occur in poorly resolved terminal or indel-associated regions. In particular, the M segment signal around position 3500 raises the possibility that W6RC.2 could represent an alternative root or near-root sequence. Because this signal involves a partially resolved insertion in a terminal non-coding region, raw-read-level validation is required before revising the outbreak rooting.
Overall, the available consensus sequences support a recent sampled genomic root close to the earliest available sequence from 26 April 2026, although this interpretation is clearest for the near-identical outbreak sequences and is complicated by the divergent Canadian consensus sequence, XDHK.2. This date reflects the root of the currently sampled sequence diversity, not the start of the outbreak, as reported cases and symptom onsets predate the first available genome sequence. The exact rooting among outbreak sequences remains uncertain pending raw-read validation of terminal differences, indel-associated patterns, and the unusually divergent Canadian sequence.
Metadata file
Hanta_metadata_outbreak_sequences.zip (2.2 KB)
Contributors to this analysis
Charu Sharma — Pandemic Sciences Institute, University of Oxford; Department of Biology, University of Oxford
Sanni Översti — Department of Biology, University of Oxford
Aris Katzourakis — Department of Biology, University of Oxford
Spyros Lytras — Antigen Evolution & Design Lab, Department of Structural Biology and Chemistry, Institut Pasteur, Paris, France
Mahan Ghafari — Pandemic Sciences Institute, University of Oxford; Department of Biology, University of Oxford