Preliminary analysis of Orthohantavirus andesense virus sequences from a cruise-ship related cluster, May 2026.
In May 2026, an Andes virus (ANDV) outbreak occurred aboard the Dutch-flagged cruise ship MV Hondius. The ship initially departed from Ushuaia, Argentina, on 1 April 2026 for a transatlantic voyage via remote South Atlantic islands. The index case (case 1) reportedly developed fever, headache, stomach pain and diarrhoea on 6 April and died onboard on 11 April, without indication of cause. During a scheduled stop in Saint Helena on 24 April, approximately 30 passengers disembarked including the wife (case 2) of index case. Most of the disembarked passengers continued their travel to Johannesburg, South Africa and then to other destinations. The body of case 1 was also removed from the ship in Saint Helena. Case 2 passed away shortly after collapsing at the airport in Johannesburg, without indication of cause. Another passenger (case 3), who fell ill since 22 April, was evacuated for medical treatment to Johannesburg on 29 April. On 2 May, case 3 tested positive for hantavirus RNA in blood and serum samples. On 3 May, case 2 tested positive for hantavirus RNA in blood and serum samples. During the first of week of May, genomic sequencing at the NICD and testing of additional passengers and crew using virus-specific PCR confirmed ANDV infection, the only hantavirus known to sustain limited human-to-human transmission, prompting public health responses to contain the outbreak, including medical evacuations and international contact tracing. As of 10 May 2026, eight cases (six confirmed and two probable) have been reported. The timeline of the cases is shown in Figure 1.
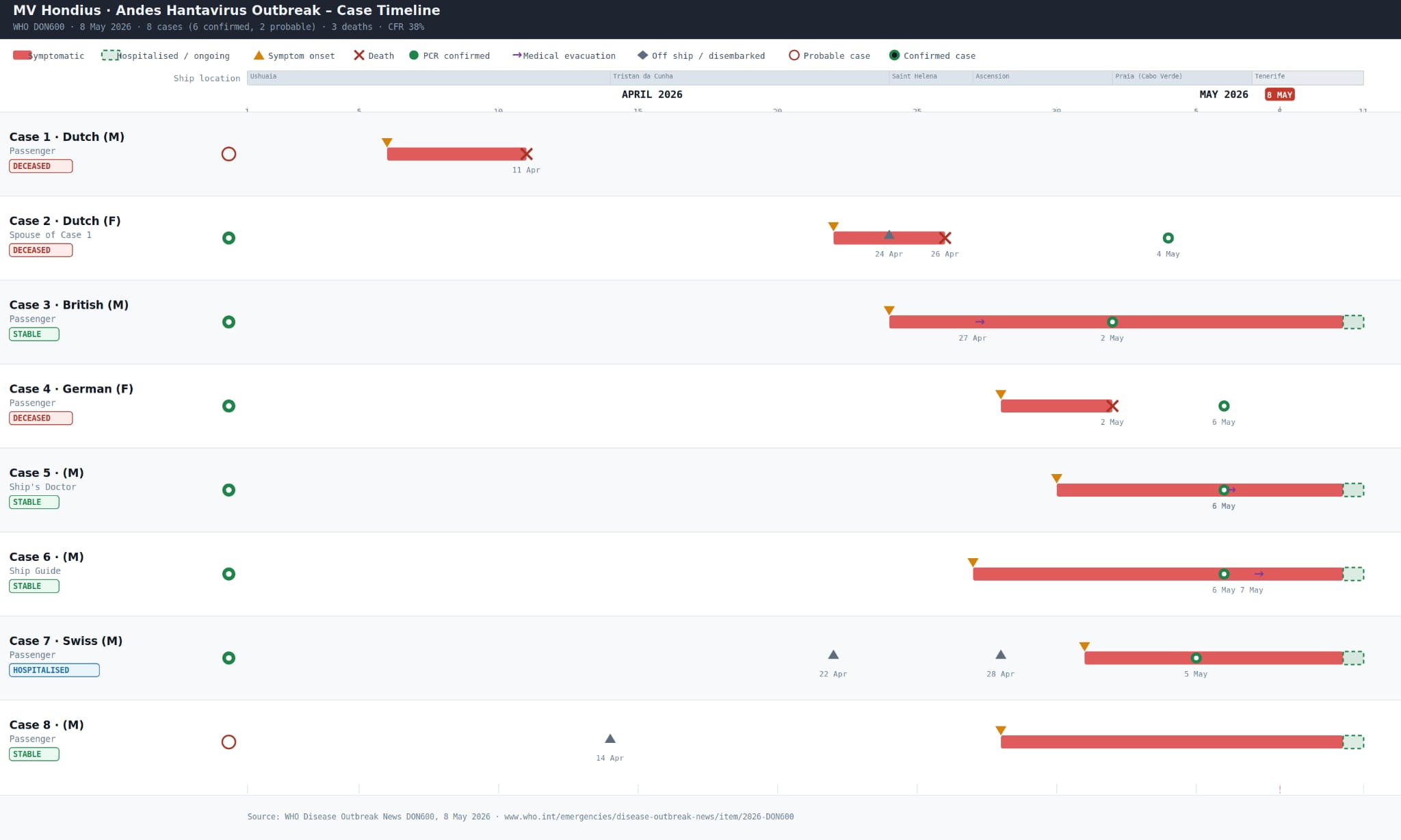
Figure 1: Timeline of the hantavirus positive cases from most of which sequence data has been generated by 10 May 2026.
Rapid review of protocols and analysis methods for quality control purposes to ensure comparability of data generated:
Because these suspected and confirmed infections were managed across several countries, clinical samples were collected, tested, and sequenced at multiple laboratories, including institutes in South Africa, Senegal, Switzerland, and the Netherlands. On 5 May, initial partial genome sequencing of the L-segment from case 2 in Johannesburg and a strain specific RT-PCR in Geneva from case 7 confirmed the presence of ANDV. On May 8th the full genome from case 7, who disembarked April 22nd at St Helena and flew back to Switzerland (from 27-28 April, via South Africa and Qatar), was shared (https://virological.org/t/complete-sequence-of-orthohantavirus-andesense-virus-swiss-resident-2026/1023). At the same date, the full genome of case 6 was also generated, and its submission to Virological and Pathoplexus is currently pending. In the context of a multi-national outbreak investigation, the participating laboratories were convened early to exchange data and assess how differences in sequencing platforms, bioinformatic pipelines, and parameter settings could affect consensus-level nucleotide polymorphisms. Different sequencing strategies were applied, including sequence-independent single-primer amplification (SISPA), unbiased metagenomics and a TWIST capture-based enrichment approach, using Illumina, Element BioSciences and Oxford Nanopore sequencing platforms (Table 1).
Table 1: Overview of the different methods used

In addition, multiple bioinformatic analysis tools were used, including de novo assembly or reference-based alignments with non-standardized sets of reference sequences. As the sequence data can be informative about the origin(s) and transmission chains, it was decided that urgent assessment of potential technical issues with the sequence data was needed. The consensus sequences and the raw sequencing reads (viral reads only) mapping to Andes orthohantavirus were shared within the group.
This process was also integrated and shared in real-time between the data producers. This coordinated process allowed the group to distinguish SNPs considered genuine from those potentially attributable to technological or analytical artifacts, thereby strengthening confidence in the genomic interpretation during the outbreak response. Although initial comparison suggested several SNPs in the sequences, this side-by-side review with a standardised set of reference sequences and agreed parameter settings resolved most of the inconsistencies. The remaining SNPs are considered true SNPs.
Comparison of consensus sequences:
Sequences from all S and M segments are identical to each other. The sequences from the L segment showed 2 SNPs: one for case 5 (position C2139T based on reference sequence MN258159) and one for case 7 (position G576A based on reference sequence MN258159) which were labelled as true SNPs rather than difference arising from technological or analytical variation. Both SNPs were synonymous mutations. All segments have high sequence identity (98.76% and 98.75% based on the L segment, 98.71% and 98.68% on the M segment and 98.73% and 98.73% on the S segment) to Andes virus sequences detected in humas in Argentina in 1997 and 2018 respectively.
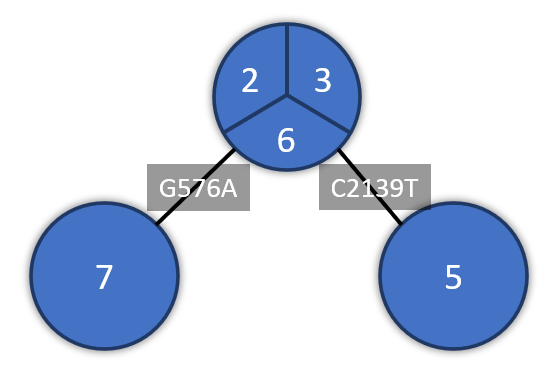
Figure 2: Minimum spanning tree of the L segment of the hantavirus positive cases for which sequence data has been generated. SNP positions are relative to reference MN258159.
The overall high level of genetic similarity — with a maximum of one detected SNP per individual — strongly suggests that the outbreak most likely originated from a single zoonotic spillover event, or a very limited number of closely related spillover events. The limited and consistent variation observed in the L segment is interpreted as true viral mutations rather than methodological artifact. Taken together, these findings support a scenario of initial zoonotic introduction followed by subsequent human‑to‑human transmission during the outbreak. The lack of diversity observed in the outbreak is similar to that observed during a cluster of human-to-human transmission in the Epuyén 2018 outbreak, in Argentina. The genomic data cannot exclude the possibility that the initial environmental exposure involved more than one passenger infected from the same source. Resolving this will require additional epidemiological data, including timelines and contact/exposure histories, together with environmental investigations such as rodent trapping and testing.
At the time of this report, sequencing efforts continue to resolve gaps and complete the analysis associated with these early cases. It is important to note that case 2, the earliest case for which a sample was available to the group, has the lowest coverage in our analysis. The sample volume was limited, and the viral load of the sample suspected to be low. Therefore, as is typical in investigations of this nature, these findings should be considered a representation of our current assessment and may be updated as additional data become available. No samples were available for case 1.
The high overall similarity among the sequences generated from different cases also supports the conclusions from the initial Swiss sequence report regarding the closest available ancestor of the outbreak virus. This conclusion may be refined as additional ANDV genomes from recent human cases, rodent reservoirs, and relevant geographic regions become available.
Moreover, this also includes sequencing of additional recent cases from the Americas to look for potential geographic clustering. Updated results are shared as they become available, and the interpretation of the data will expand accordingly. In addition, the aim was to incorporate any available reliable sequence data, despite variable quality of samples and for some cases low viral loads. Therefore, additional analyses are ongoing, but this data is released to support ongoing public health purposes. Updated analysis will continue to be shared as it becomes available.
Summary:
Genomic sequencing has become a standard to support outbreak response activities. The rapid identification of ANDV from partial genome sequences provided by NICD, and the release of the first full genome by Geneva University Hospitals and University of Zürich, within days after identifying the outbreak, provided important information for initial assessment of the etiology and the consequences of that for choice of diagnostic detection methods. The necessary multi-partner quality control process organised rapidly and was intended to allow for release of further sequence data that is directly actionable. We propose to continue this process in the case of additional data generated, to ensure high quality genomic support.
Data availability:
The consensus sequences are shared on the Pathoplexus platform under the following accession numbers:

Contact persons:
Bas Oude Munnink ([email protected])
Michael Huber ([email protected])
Samuel Cordey ([email protected])
Moussa Moise Diagne ([email protected])
Jacqueline Weyer ([email protected])
Report prepared by (in alphabetic order by first name):
Persons involved in diagnostics, sequencing, data contribution and analysis, and interpretation (in alphabetical order by first name):

References
Alonso, D.O., Pérez-Sautu, U., Bellomo, C.M., Prieto, K., Iglesias, A., Coelho, R., Periolo, N., Domenech, I., Talmon, G., Hansen, R. and Palacios, G., 2020. Person-to-person transmission of Andes virus in hantavirus pulmonary syndrome, Argentina, 2014. Emerging infectious diseases, 26(4), p.756.
Alonso, D.O., Kehl, S.D., Coelho, R.M., Periolo, N., Poklépovich Caride, T., Sanchez Loria, J., Cuba, F.G., Pérez-Sautu, U., Sanchez-Lockhart, M., Palacios, G. and Bellomo, C.M., 2024. Orthohantavirus diversity in Central-East Argentina: Insights from complete genomic sequencing on phylogenetics, Geographic patterns and transmission scenarios. PLoS neglected tropical diseases, 18(10), p.e0012465.
Bellomo, C.M., Kehl, S., Alonso, D.O., López, W., Cassinelli, F., Coelho, R.M., Bravo, G., Aguirre, S., Dib, M., Periolo, N. and Toscano, C., 2025. Novel Orthohantavirus Associated with Hantavirus Pulmonary Syndrome in Northern Argentina. Viruses, 17(5), p.717.
Ferrés M, Martínez-Valdebenito C, Henriquez C, Marco C, Angulo J, Barrera A, Palma C, Barriga Pinto G, Cuiza A, Ferreira L, Rioseco ML, Calvo M, Fritz R, Bravo S, Bruhn A, Graf J, Llancaqueo A, Rivera G, Cerda C, Tischler N, Valdivieso F, Vial P, Mertz G, Vial C, Le Corre N. (2024). Viral shedding and viraemia of Andes virus during acute hantavirus infection: a prospective study. Lancet Infect Dis. S1473-3099(24)00142-7.
Martínez, V.P., Di Paola, N., Alonso, D.O., Pérez-Sautu, U., Bellomo, C.M., Iglesias, A.A., Coelho, R.M., López, B., Periolo, N., Larson, P.A. and Nagle, E.R., 2020. “Super-spreaders” and person-to-person transmission of Andes virus in Argentina. New England Journal of Medicine, 383(23), pp.2230-2241.
Song, D.H., Kim, W.K., Gu, S.H., Lee, D., Kim, J.A., No, J.S., Lee, S.H., Wiley, M.R., Palacios, G., Song, J.W. and Jeong, S.T., 2017. Sequence-Independent, Single-Primer Amplification Next-Generation Sequencing of Hantaan Virus Cell Culture–Based Isolates. The American journal of tropical medicine and hygiene, 96(2), p.389.
World Health Organization, Hantavirus cluster linked to cruise ship travel, Multi-country (8 May 2026), https://www.who.int/emergencies/disease-outbreak-news/item/2026-DON600 (Accessed on 10 May 2026)