Context
The G21987A mutation, which is found in the delta lineage of SARS-CoV-2, is often absent from consensi generated at the Health2030 Genome Center (Geneva), which led to the classification of the samples as AY* sublineage. However, the consensi generated at the Centre Hospitalier Universitaire Vaudois (CHUV) contain the mutation, showing a discrepancy between our 2 centres (Trestan Pillonel, CHUV, personal communication)
The protocol used at CHUV for sequencing SARS-CoV-2 is based on the PARAGON GENOMICS CleanPlex primer set, whereas we generate amplicons based on ARTIC v3 (Illumina COVIDSeq). In fact, it has recently been described by Alban Ramette’s lab (Institute for Infectious Diseases, IFIK) that the 21300-22300 region is not correctly amplified by the ARTIC v3 primer set (https://www.medrxiv.org/content/10.1101/2021.09.09.21262951v1.full-text).
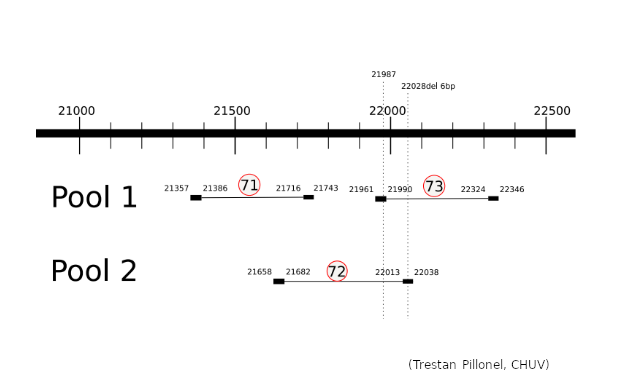
The Illumina COVIDSeq amplification protocol uses 2 separate primer pools generating non-overlapping amplicons that are pooled in the following steps of the procedure to cover the entire genome. To understand why we do not observe the G21987A mutation, we prepared samples of the SARS-CoV-2 alpha (4 samples) and delta (4 samples) variants (courtesy provided by Hôpitaux Universitaires de Genève, HUG) using the Illumina COVIDSeq protocol, but sequenced the 2 pools separately. We focused on the region of interest, pos 21300-22300 of the SARS-CoV-2 genome, which is covered by 3 amplicons produced by ARTIC v3 primers.
Observations for alpha variants
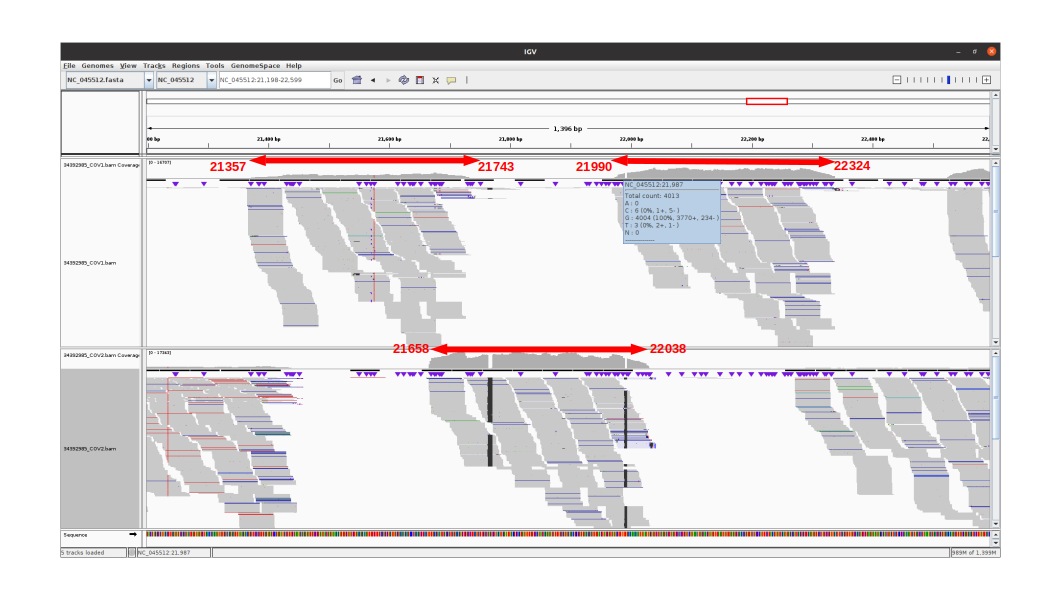
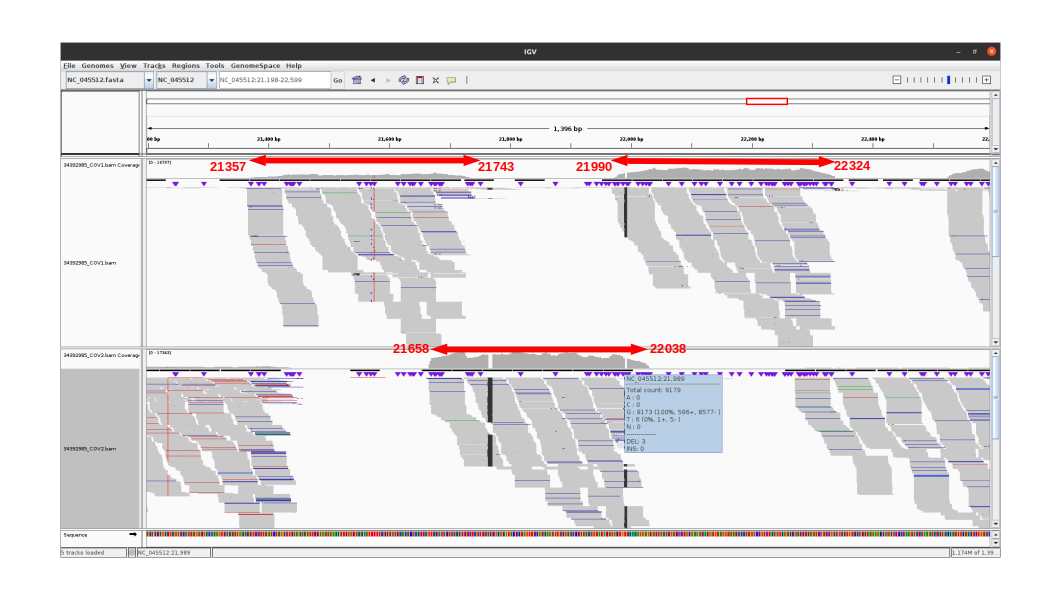
The 3 amplicons of interest are correctly amplified. The coverage at position 21987 for pool 1 (mean=3931.3 std=526.2) is lower than for pool 2 (mean=10545.8 std=2188.7), but within the ranges expected by our protocol.
An IGV view of the region of interest for one alpha variant analyzed:
Observations for delta variants
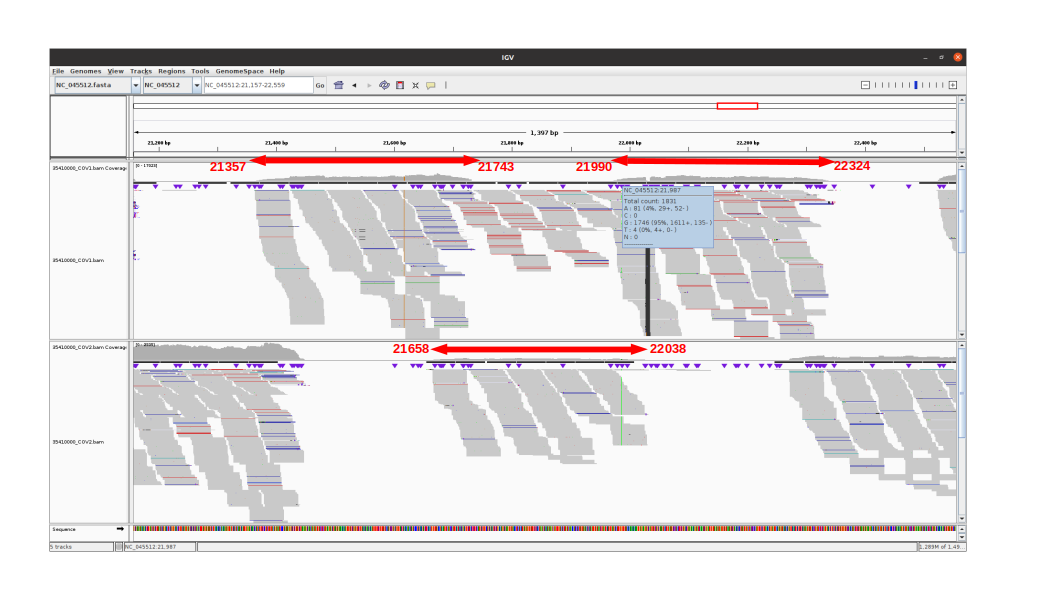
Amplicon 21990-22324 is not efficiently amplified (coverage at pos 21987: mean=2224.0, sdt=1393.9), and the majority of the reads do not come from the amplicon itself, but from amplification between this amplicon and the previous one in the pool. Our hypothesis is that 1) at the initial PCR cycles there is some kind of suboptimal amplification of the 2 expected regions and 2) in the later stages of the PCR there is an amplification between the 2 produced amplicons, probably due to better affinity than the original primers. This would result in the integration of the G present in the original primer in the reads.
The 21658-22038 amplicon in pool 2 is barely amplified (coverage at pos 21987: mean=167.8, std=86.4), but includes the expected nucleotide A.
An IGV view of the region of interest for one delta variant analyzed:
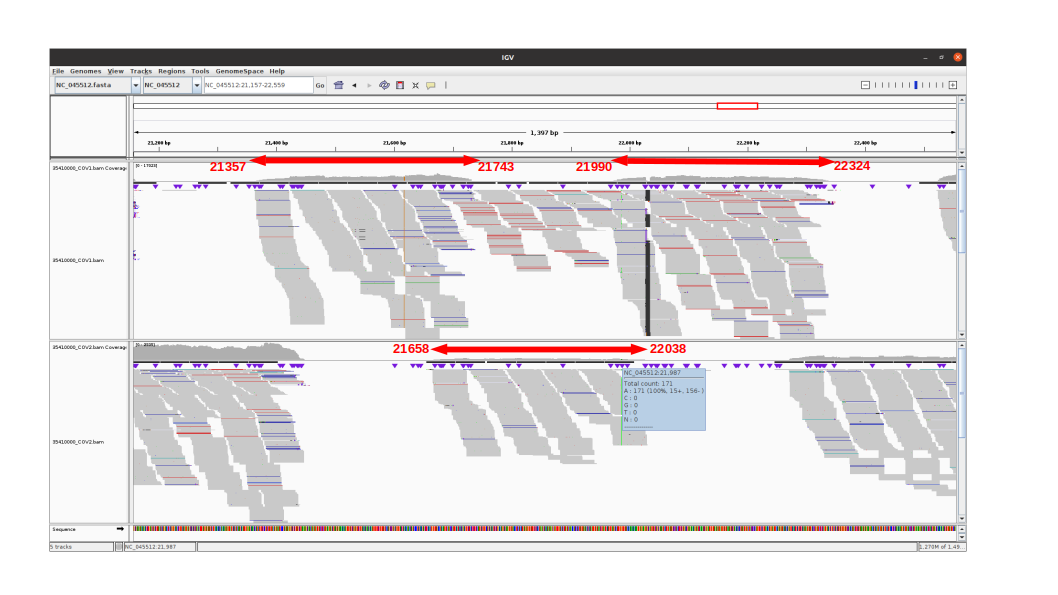
Conclusion
The G21987A mutation is mostly absent from the sequencing of the SARS-CoV-2 delta lineage because the ARTIC v3 protocol produces a non-specific amplification between amplicons 21357-21743 and 21990-22324 which would incorporate the G at position 21987 from the primer sequence. This is not counterbalanced by amplicon 21658-22038 as it shows consistently a much lower amplification. The 5’ primer anchor of the 21990-22324 amplicon would be lost due to non-specific amplification, hence the ineffectiveness of standard tools in removing primers from reads in this region.
The problem described should be corrected by the ARTIC v4 version of the primers, which will soon be available as an add-on to the Illumina COVIDSeq kit (Illumina, personal communication).
