Systematic Errors Associated with Some Implementations of ARTIC V4 and a Fast Workflow to Prescreen Samples for New Problematic Sites
Theo Sanderson1,2,* , Nicola De Maio3, Angie S. Hinrichs4, Adriano de Bernardi Schneider4,5, Conor Walker3, Nick Goldman3, Yatish Turakhia 6, Robert Lanfear 7, Russell Corbett-Detig4,5,*
1Francis Crick Institute, London, UK
2Wellcome Sanger Institute, Hinxton, Cambridgeshire, UK
3European Molecular Biology Laboratory, European Bioinformatics Institute, Hinxton, Cambridgeshire, United Kingdom
4Genomics Institute, University of California Santa Cruz, Santa Cruz, CA, USA
5Department of Biomolecular Engineering, University of California Santa Cruz, Santa Cruz, CA, USA
6Department of Electrical and Computer Engineering, University of California San
Diego, San Diego, CA, USA
7Department of Ecology and Evolution, Research School of Biology, Australian National University, Canberra, ACT, Australia
* [email protected], [email protected]
Systematic Errors Associated with Some Implementations of ARTIC V4
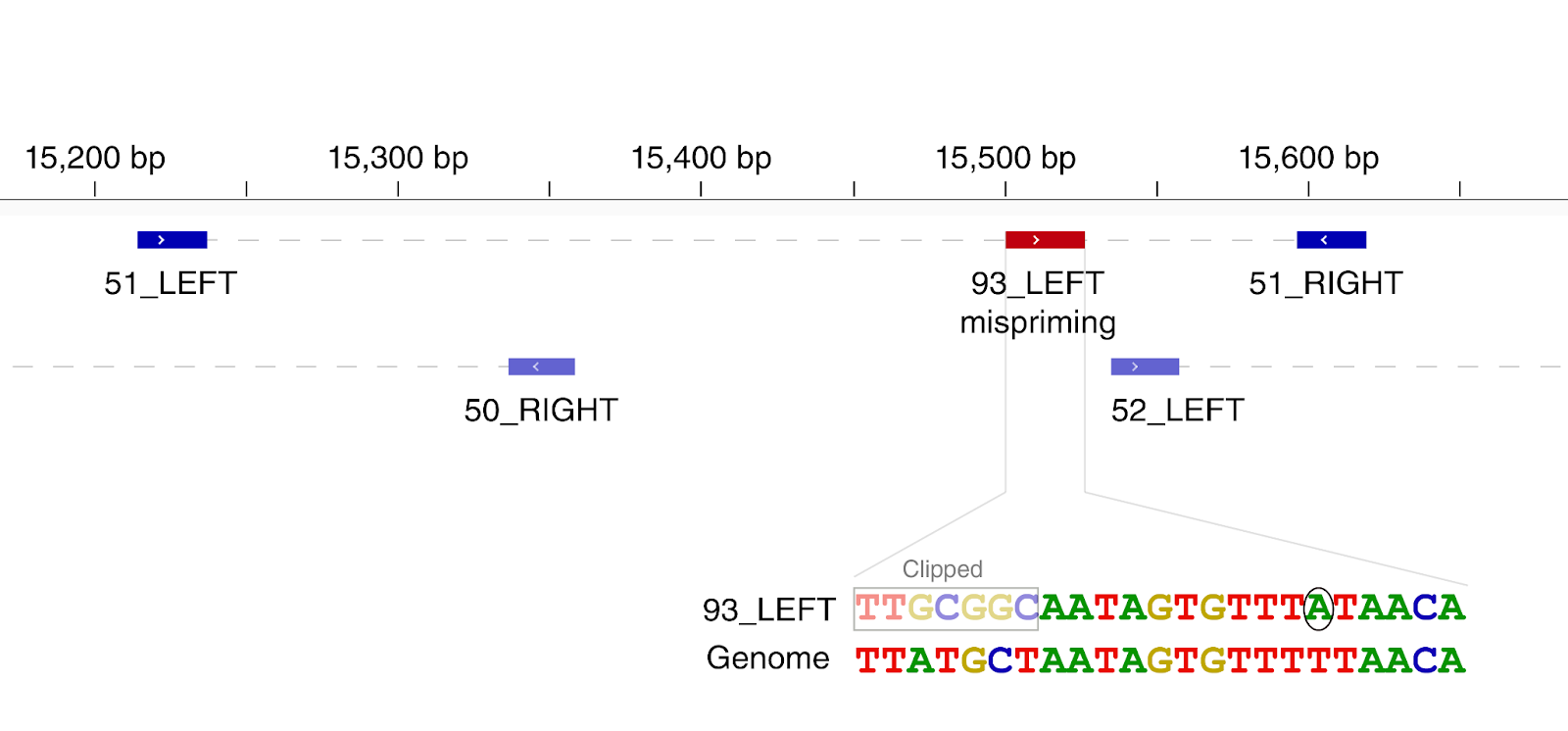
The ARTIC V4 primers [1] are an important update to the primer scheme that has driven most sequencing during the SARS-CoV-2 pandemic. V4 resolves a number of amplification difficulties in ARTIC V3 introduced by mutations in SARS-CoV-2 variants of concern, including Delta. However, two genomic artifacts have been observed in substantial quantities from some implementations of the ARTIC V4 primers. Primer v4:93_LEFT, in addition to its intended binding site, also shows homology to a region within amplicon 51 where it can prime a secondary amplicon (in a pair with correctly-binding primer v4:51_RIGHT). Due to a difference between the primer and the region to which it binds, this creates the appearance of a T15521A mutation, which would encode orf1ab:F5086Y (a.k.a. ORF1b:F685Y). Similarly, primer v4:41_LEFT shows additional homology to a region within amplicon 29, where it can prime a secondary amplicon (in a pair with correctly-binding primer v4:29_RIGHT), amplifying a truncated version of amplicon 29 that incorporates an apparent T8835C SNP, from a mismatch within the primer, creating the appearance of an orf1ab:V2857A mutation. In each case, a 5’ mismatching region of the primer is softclipped during mapping. Since these are not standard primer sites, these primer-induced artefacts will survive basic primer trimming.
Figure 1. Mispriming with v4:93_LEFT at a region within amplicon 51 can create a systematic error. The apparent T15521A substitution created by the presence of this primer-derived sequence is circled. Odd and even numbered primers are used in distinct pools.
Solutions to avoid this issue will depend on the workflow used. For sequencing chemistries where one read – or one read-pair – corresponds to an entire amplicon, filtering based on insert-size, or on the presence of canonical primer sequences at both ends, will be helpful. Such an approach is already implemented in the ARTIC Network’s pipeline for nanopore sequencing, where guppyplex filters on amplicon size and align_trim filters on the presence of correct primers. The most challenging case to solve is where amplicons are fragmented prior to short-read sequencing, given that the misprimed amplicon that truncates amplicon 29 has a length >200bp. Removing soft-clipped reads may be of some assistance here. In the longer term, future primer schemes will be screened to avoid potential mispriming sites. These issues, and others observed in some implementations of the V3 primers [2–4], highlight the need for vigilance against technical artefacts that can distort phylogenetic trees.
Due to the frequency at which T15521A and T8835C errors are currently observed, we have added these positions to our masking recommendations for SARS-CoV-2 alignments: problematic_sites_sarsCov2.vcf.
A Workflow for Prescreening New Samples for Systematic Errors
We developed a workflow that can be used to prescreen samples for previously undescribed possible systematic errors. Briefly, the idea is to add newly sequenced samples to the public tree that we maintain [5], and to identify specific variants at individual sites whose parsimony score increased the most as a consequence of adding the new samples. This might be useful for users when evaluating samples produced on a new sequencing run, new wet lab or bioinformatic protocols, or a new batch of sequencing or library preparation reagents. This workflow is intended to facilitate identification of potential systematic errors or otherwise problematic positions in the genome, but it is not a definitive analysis. The results should be accompanied by detailed interpretation of the specific sites that the workflow identified.
To demonstrate the utility of this workflow, we recreated the conditions where it could be used to flag the likely systematic errors that appear to be associated with the new ARTIC V4 primer set. To do this, we pruned the approximately 10,000 samples sequenced at the Wellcome Sanger Institute using the V4 primer scheme that had already been included in the public UShER mutation annotated tree from 10/28 [5]. The idea is that we can recreate a use case for this workflow by analyzing this set of samples. We then analysed all the pruned sequences found in the current COG-UK dataset: aligning them to the reference (NC_045512.2), masking previously known problematic sites, and placing them onto this pruned tree. Exact commands to reproduce this analysis are available from this github repository. Running this workflow identified the likely problematic positions as specific variants at individual sites whose parsimony score increased the most as well as apparent back mutations at those same positions after adding the affected samples.
| variant | parsimony_public | parsimony_total | parsimony_user | parsimony_public/sample | parsimony_user/sample |
|---|---|---|---|---|---|
| T15521A | 1244 | 3246 | 2002 | 0.002629475199 | 0.4041922989 |
| T8835C | 880 | 1586 | 706 | 0.0005531057063 | 0.2280701754 |
| A15521T | 10 | 100 | 90 | 0.0003912644868 | 0.08042834359 |
| G72A | 22 | 97 | 75 | 4.45E-06 | 1.03E-02 |
| G22032T | 56 | 128 | 72 | 9.78E-06 | 8.54E-03 |
| A22030G | 1 | 72 | 71 | 2.49E-05 | 8.20E-03 |
| A26766C | 16 | 81 | 65 | 4.45E-07 | 8.09E-03 |
| A28248G | 44 | 88 | 44 | 7.11E-06 | 7.40E-03 |
| C8835T | 4 | 42 | 38 | 1.96E-05 | 5.01E-03 |
| T27904G | 65 | 88 | 23 | 1.78E-06 | 4.33E-03 |
Table 1: The first ten lines of this workflow’s output includes variants whose parsimony score increased the most due to the addition of the user’s samples. Forward and reverse mutations at the two apparently problematic positions are identified.
Interpretation Guidelines
Here we provide some basic guidelines for using this workflow to prescreen newly produced data. Although our workflow can help to identify problematic positions, it may not be obvious what the source of apparently high parsimony score variants at a specific site is. We caution that many factors may influence the resulting parsimony score increases. For example, the phylogenetic diversity within the dataset examined and within a local region will affect our ability to identify such apparent variants. Additionally, because other problematic sites can distort phylogenetic inference, true mutations may display apparently elevated parsimony scores when samples containing these alleles are placed incorrectly on the phylogeny.
Due to the range of factors that can affect these results, we recommend that users of this workflow treat it as only a first pass analysis. Users may wish to run the workflow, identify the variant that generates parsimony score increase and examine the short read alignment files underlying such positions. If this appears to be a systematic error, users should update their workflow to repair this variant call or simply mask the position and rerun the workflow on the updated sequences to determine if other sites are indeed potentially problematic.
Running the Workflow
The ideal use of this workflow would be to run it before uploading samples to sequence repositories so as to identify potential problems in the sequences before sharing them. To run this workflow on your own data, provide the sequence data as a multi-fasta file and run the following commands (dependencies conda, mamba, snakemake, git and a fasta file):
git clone https://github.com/yatisht/usher.git
cd usher/workflows/
snakemake --use-conda --cores [num_threads] --config RUNTYPE=”systematic” FASTA=”[full/path/to/fasta/file]”
The output table, parsimony_report.txt, will contain the variants whose parsimony score increased as a result of adding the user’s samples. We emphasize that this is intended only to flag potentially problematic positions and we anticipate that users may want to deeply investigate sites that have especially large impacts on the estimated parsimony score.
Exact commands to reproduce the analysis we showcased here are available via github. This is slightly different from above because we needed to extract the Sanger samples produced using the ARTIC V4 primers.
Acknowledgements
We are grateful to Moritz Gerstung, @babarlelephant, Alex Selby, Jeffrey Barrett, Rob Davies, Nick Loman and Joshua Quick for assistance with understanding ARTIC V4 issues and possible solutions. We thank the generators of sequences analysed here: the COG-UK Consortium, the Wellcome Sanger Institute COVID-19 Surveillance Team, and all submitters to the INSDC databases.
References
- SARS-CoV-2 version 4 scheme release - Laboratory - ARTIC Real-time Genomic Surveillance
- Sanderson T, Barrett JC. Variation at Spike position 142 in SARS-CoV-2 Delta genomes is a technical artifact caused by dropout of a sequencing amplicon. medRxiv. 2021; 2021.10.14.21264847.
- lcerutti, j_quick, connortr, arambaut, n_j_loman, vborges, et al. Missing G21987A mutation in SARS-CoV-2 delta variants due to non-specific amplification by ARTIC v3 primers. 20 Oct 2021. Available: https://pando.tools/t/missing-g21987a-mutation-in-sars-cov-2-delta-variants-due-to-non-specific-amplification-by-artic-v3-primers/764
- Davis JJ, Wesley Long S, Christensen PA, Olsen RJ, Olson R, Shukla M, et al. Analysis of the ARTIC version 3 and version 4 SARS-CoV-2 primers and their impact on the detection of the G142D amino acid substitution in the spike protein. bioRxiv. 2021; doi:10.1101/2021.09.27.461949
- McBroome J, Thornlow B, Hinrichs AS, Kramer A, De Maio N, Goldman N, et al. A daily-updated database and tools for comprehensive SARS-CoV-2 mutation-annotated trees. Mol Biol Evol; 2021. doi:10.1093/molbev/msab264