Increasing frequency of SARS-CoV-2 lineages B.1.1.7, P.1 and P.2 and identification of a novel lineage harboring E484Q and N501T spike mutations in Minas Gerais, Southeast Brazil
Filipe Romero Rebello Moreira 1*, Diego Menezes Bonfim 2*, Victor Emmanuel Viana Geddes 2*, Danielle Alves Gomes Zauli 3, Joice do Prado Silva 3, Aline Brito de Lima 3, Frederico Scott Varella Malta 3, Alessandro Clayton de Souza Ferreira 3, Victor Cavalcanti Pardini 3, Daniel Costa Queiroz 2, Rafael Marques de Souza 2, João Locke Ferreira de Araújo 2, Hugo José Alves 2, Ana Valesca Fernandes Gilson Silva 4, Gustavo Gomes Resende 5, André Luiz de Menezes 6, Eneida Santos de Oliveira 6, Jaqueline Silva de Oliveira 7,Mauro Martins Teixeira 8, Lucyene Miguita Luiz 9, Ricardo Santiago Gomez 10, Paula Luize Camargos Fonseca 2, Rennan Garcias Moreira 11, Amilcar Tanuri 1, William Marciel de Souza 12, Nuno Rodrigues Faria 13,14,15, Carolina Moreira Voloch 1**, Renan Pedra de Souza 2**, Renato Santana Aguiar 2,16**![]()
*These authors share the first authorship. **These authors share the senior authorship.
![]() Correspondence: R. S. Aguiar (ORCID: 0000-0001-5180-3717. Contact: [email protected])
Correspondence: R. S. Aguiar (ORCID: 0000-0001-5180-3717. Contact: [email protected])
1 - Laboratório de Virologia Molecular, Departamento de Genética, Instituto de Biologia, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil (F.R.R. Moreira, C.M. Voloch, A. Tanuri)
2 - Laboratório de Biologia Integrativa, Departamento de Genética, Ecologia e Evolução, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte, Brasil (D.M. Bonfim, V.E.V. Geddes, D.C. Queiroz, R.M. Souza, P.L.C. Fonseca, J.L.F. Araújo, H.J. Alves, R.P. Souza andR.S. Aguiar)
3 - Instituto Hermes Pardini, Belo Horizonte, Brazil (D.A.G. Zauli, J.P. Silva, A.B. Lima, F.S.V. Malta, A.C.S. Ferreira, V.C. Pardini)
4 - Escola de Saúde Pública de Betim, Secretaria Municipal de Saúde, Prefeitura de Betim, Betim, Brasil (A.V.F.G. Silva)
5 - Reumatologia, Hospital das Clínicas, Empresa Brasileira de Serviços Hospitalares, Universidade Federal de Minas Gerais, Belo Horizonte, Brasil (G.G. Resende)
6 - Secretaria Municipal de Saúde, Prefeitura de Belo Horizonte, Belo Horizonte, Brazil (A.L. de Menezes, E.S. de Oliveira)
7 - Laboratório e Pesquisa em Vigilância (CELP), Secretaria de Estado de Saúde de Minas Gerais (SES-MG) (J.S. Oliveira)
8 - Departamento de Bioquímica e Imunologia, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte, Brasil (M.M. Teixeira)
9 - Departamento de Patologia, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte, Brasil (L.M. Luiz)
10 - Departamento de Cirurgia Oral e Patologia, Faculdade de Odontologia, Universidade Federal de Minas Gerais, Belo Horizonte, Brasil (R.S. Gomez)
11 - Centro de Laboratórios Multiusuários, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte, Brasil (R.G. Moreira)
12 - Virology Research Centre, Ribeirão Preto Medical School, University of São Paulo, Ribeirão Preto, SP, Brazil (W.M. Souza).
13 - Department of Infectious Disease Epidemiology, Imperial College London, London, UK (W.M. Souza, N.R. Faria)
14 - Instituto de Medicina Tropical, Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brasil (N.R. Faria)
15 - Department of Zoology, University of Oxford, Oxford, UK (N.R. Faria)
16 - Instituto D’Or de Pesquisa e Ensino (IDOR), Rio de Janeiro, Brasil (R.S. Aguiar)
Summary
We report preliminary results of an ongoing investigation of SARS-CoV-2 genomic diversity in the metropolitan region of Belo Horizonte (MRBH), Minas Gerais, Brazil. We sequenced and characterized 85 nearly complete SARS-CoV-2 genome sequences from randomized samples collected between 28 October 2020 and 15 March 2021. Phylogenetic analysis reveals co-circulation of two variants of concern (VOC), B.1.1.7 (n=3, 3.53%) and P.1 (n=30, 35.29%), and variant of interest (VOI) P.2 (n=41, 48.23%). These variants harbor E484K (P.1 and P.2) and N501Y (P.1 and B.1.1.7) mutations that are associated with increased transmissibility or immune escape. The N501Y mutation has also been associated with an increase in COVID-19 hospitalizations and deaths. Notably, we find that between 28 Feb and 15 Mar, 68% of cases were caused by the P.1 lineage in the MRBH. In addition, we report a cluster of two sequences characterized by a unique array of 18 mutations, including new non-synonymous changes in the same critical spike amino acid positions, E484Q and N501T. This lineage seems to have emerged independently from the nationally widespread B.1.1.28, as previously reported for P.1 and P.2, and adds up to the composition of a complex epidemiological scenario of the SARS-CoV-2 pandemic in Brazil.
Context
Over the last months, several teams have reported the emergence of multiple variants of concern (VOC) or interest (VOI) in Brazil, such as P.1 (1), P.2 (2), N.9 (3), N.10 (4) and B.1.351 (5), documenting their geographical dissemination throughout the country (6). While the epidemiological consequences of their co-circulation dynamics remain unclear, current evidence supports that P.1, B.1.1.7 and B.1.351 are associated with altered epidemiological characteristics (1, 7, 8). These VOCs harbor unique constellations of mutations that are associated with increased transmissibility (1, 7, 8), immune escape (9) and/or increased disease severity (1, 10).
In a highly complex epidemiological scenario (11, 12), we sought to determine the frequency of SARS-CoV-2 variants in the metropolitan region of Belo Horizonte, the capital city of Minas Gerais state, southeast Brazil. Although Minas Gerais is the second most populous Brazilian federal unit (~21.2 million individuals), the diversity of recently described viral lineages circulating in this region remains poorly understood. Here, we characterize 85 SARS-CoV-2 genome sequences from Minas Gerais, including 78 collected from January 7th to March 15th, 2021.
Study
Sampling strategy and genome sequencing
To ascertain the current scenario of the COVID-19 pandemic in the MRBH, we conducted a genomic surveillance study based on 85 randomly selected clinical samples positive for SARS-CoV-2, collected between 28th October 2020 and 15th March 2021. Samples were collected in three laboratories (Laboratório Hermes Pardini, Laboratório de Biologia Integrativa, UFMG, and Laboratório Municipal de Referência, PBH) and molecular diagnostics was conducted with RT-qPCR, using either CDC N1 and N2 primers (13) or the TaqPath COVID-19 assay (Thermo Fisher, USA). Sequencing libraries were prepared using the QIAseq FX DNA Library Prep kit (QIAGEN, Germany) and sequenced on the Illumina MiSeq platform (Illumina, USA) with a v3 (600 cycles) cartridge. Three negative controls were used in all sample processing steps (cDNA synthesis, viral genome amplification and library preparation). Quality control and consensus genome sequence inferences were performed with a custom pipeline released in the context of a previous study performed by our group (14). All mutations detected in the novel consensus genome sequences and reported in this study were manually verified in raw sequencing data. Sequencing statistics and metadata for samples are available in Supplementary Table 1.|
Sequencing throughput corresponded to approximately 55,8 million reads. All sequenced samples had genome coverage above 70% (mean 10x coverage: 97.33% range: 72.16 - 99.89%; mean 100x coverage: 90.37%, range: 51.45 - 99.77%; mean depth: 2298x, range: 127 – 11,487). Even though average sequencing depth was unbalanced among samples, 72 (84.71%) of them have been sequenced at least 500x and all above 100x (Supplementary Table 1). Negative controls were clear.
Genomic analysis reveals co-circulation of lineages P.1 and B.1.1.7 in Belo Horizonte
After manual inspection of mutations identified, the sequences were classified using the Pangolin tool (15), as: P.1 (30, 35.29%), P.2 (41, 48.23%), B.1.1.28 (8, 9.41%), B.1.1.7 (3, 3.53%), B.1.1.143 (1, 1.17%), B.1.235 (1, 1.17%) and B.1.1.94 (1, 1.17%). This is the first detection of lineage B.1.1.7 from randomly selected samples (instead of samples presenting S gene target failure on RT-qPCR) in Brazil, suggesting this lineage is becoming epidemiologically relevant in the country (14).
Despite our limited sample size, we were able to detect a change in the genetic composition of the SARS-CoV-2 epidemic population in the MRBH. While P.2 had been the major lineage through most of the study period, the relative frequency of P.1 has rapidly increased since its first detection in our data (February 24, 2021), becoming the dominant lineage in the following weeks (Figure 1). Overall, our results highlight the predominance of VOC/VOI (P.1, P.2 and B.1.1.7) over previously circulating lineages in Minas Gerais, B.1.1.28 and B.1.1.33, the major strains circulating in Brazil since the beginning of the pandemic. This pattern of lineage replacement agrees with the observed in other regions of the country, adding to the increasing body of evidence suggesting relative fitness advantage of VOCs (1, 7, 8). Similar shifts from non-VOC to VOC dominance have been shown to have epidemiological consequences, including faster epidemic spread, shifts in the age distribution of infected individuals and increased mortality rates (1, 7, 8, 10, 16).
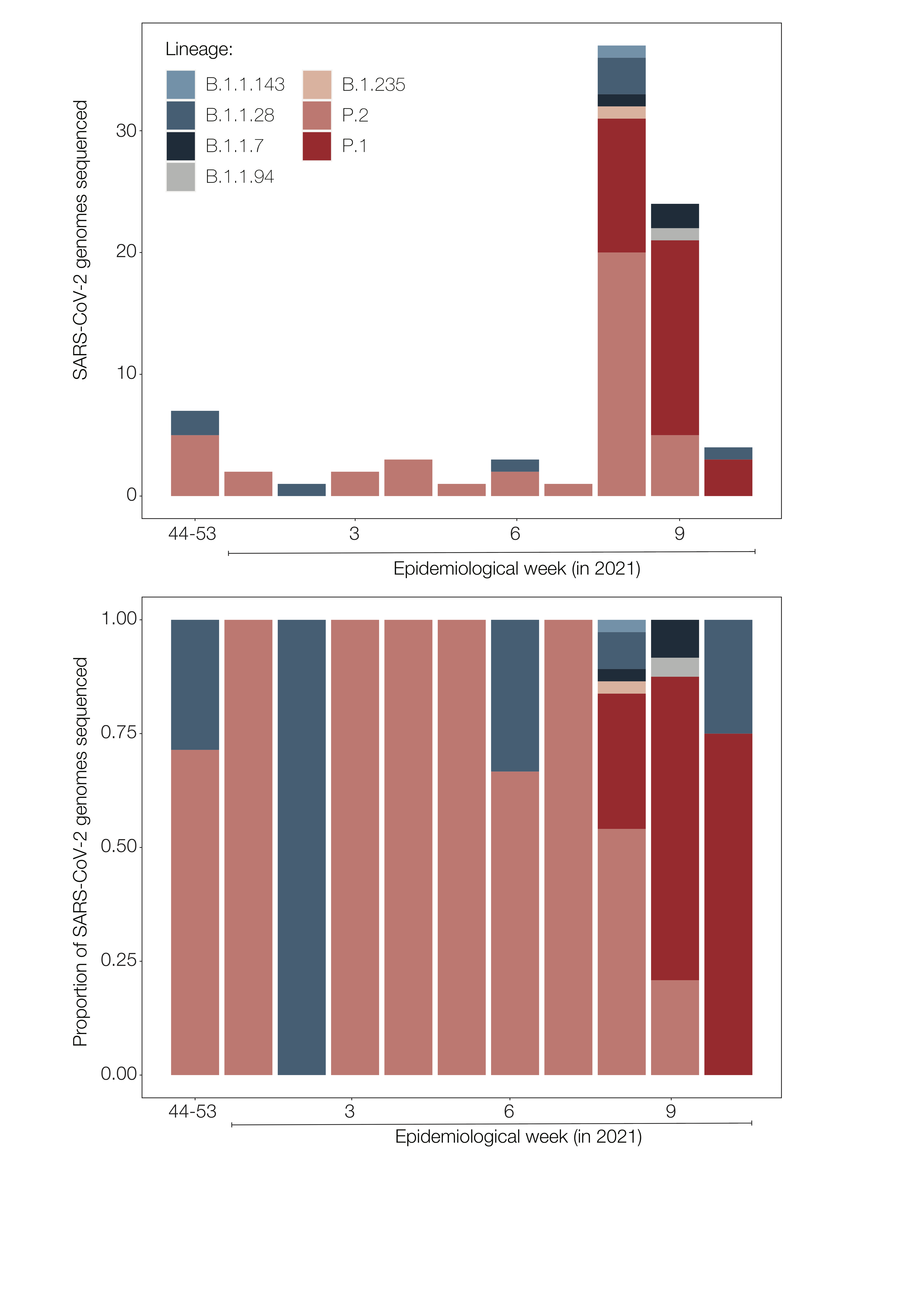
Figure 1. Barplots exhibiting the variation in lineages’ frequencies in Minas Gerais state along epidemiological weeks of 2020 and 2021. The upper panel depicts the temporal distribution of lineages in absolute numbers of SARS-CoV-2 genome sequences available, while the lower exhibit relative frequencies. The plots reveal that the epidemiological dynamics of SARS-CoV-2 in this region has been dominated by the circulation of variants of interest (P.2) or concern (P.1). Specifically, P.2 circulated in high frequency along the eight first epidemiological weeks of 2021, a scenario that changed with the arrival and spread of lineage P.1, which became the major lineage over the following weeks.
To further contextualize the genetic diversity of SARS-CoV-2 circulating in the MRBH, we assembled a comprehensive and globally representative dataset of genome sequences available on GISAID (n = 3,273). This dataset was composed by randomly sampling half the sequences of a larger dataset assembled from all Brazilian sequences available and sampled international sequences (one per country per epidemiological week). A maximum-likelihood tree was inferred from this dataset with IQ-Tree 2 (17) under the GTR+F+I+G4 model (18, 19) and Shimoidara-Hasegawa-like approximate likelihood ratio test (SH-aLRT; 20) was used to assess branches’ statistical support. Our phylogenetic reconstruction confirmed pangolin classification for most genome sequences and identified the co-circulation of multiple VOC/VOI in the MRBH during the sampled period (Figure 2). Nevertheless, six sequences identified phylogenetically as P.1 (n=1), and P.2 (n=5) were classified as B.1.1.28 in pangolin; and one phylogenetically identified B.1.1.28 genome was classified as B.1.1.143 in pangolin. New sequences from Belo Horizonte cluster with sequences from several states scattered in Brazilian regions, emphasizing the role of human mobility as a driver of viral dissemination. Despite this general pattern, we find 13 monophyletic clades majorly composed of sequences from the MRBH within P.1 and P.2 lineages. These clusters have variable sizes, between 2 and 15 sequences, and were inferred with 76.7 to 99 SH-aLRT statistical support. The detailed annotated maximum likelihood tree can be found here.

Figure 2. Maximum likelihood phylogeny revealing the diversity of lineages circulating in Belo Horizonte metropolitan region in the sampled period. Tip colors indicate sequence sampling location: International, Brazil or metropolitan region of Belo Horizonte (MRBH). While the majority of novel sequences belong to previously characterized VOC/VOIs (B.1.1.7, P.1 and P2), a highly supported monophyletic cluster derived from lineage B.1.1.28 and defined by 18 unique mutations have been inferred (samples LBI215 and LBI218). The inset highlights the novel described lineage and its defining mutations are listed in the associated table. As these samples share several non-synonymous mutations of putative biological significance in the spike protein (G23012C: E484Q, A23064C: N501T, C24374T: L938F, G24410A: D950N) and are not epidemiologically linked, we believe these results could indicate the existence of a novel VOI. We highlight the new N501T and E484Q substitutions at the same amino acid positions shared by B.1.1.7, B.1.351, P.1 and P.2 variants.
Genomic description of a putative novel B.1.1.28-derived VOI
Our analysis revealed the circulation of a putative new variant derived from lineage B.1.1.28, and represented by two genome sequences in our dataset, LBI215 (complete, 99.81% 10x genome coverage, 98.07% 100x genome coverage, average sequencing depth: 1532x, pangolin original classification: B.1.235) and LBI218 (partial, 76.20% 10x genome coverage, 57.45% 100x genome coverage, average sequencing depth: 595x, pangolin original classification: B.1.1.94). These two sequences form a well-supported (SH-aLRT = 100) monophyletic clade characterized by a unique set of 18 nucleotide mutations (Figure 2).
Beyond harboring multiple exclusive synonymous (C1627U, A10888G, C12664U, C24904U, C27807U, A28271U) and non-synonymous (G5180A: ORF1ab D1639N, G9929A: ORF1ab D3222N, G23012C: Spike E484Q, A23064C: Spike N501T, C24374U: Spike L938F, G24410A: Spike D950N, C28311U: Nucleocapsid P13L) SNPs, this lineage also possesses mutations present in other VOCs: deletion 11288-11296 (ORF1ab 3675-3677 SGF; shared by P.1, B.1.1.7 and B.1.351), C21614U (Spike L18F, P.1) and C28253U (Synonymous, P.2). Two additional deletions are present in these sequences: deletions 28881-28889 (Nucleocapsid 203-206 RGTS(T)) and 29581, which causes a frameshift mutation in ORF10. Additional mutations covered only in the LBI215 genome include: G3617A, ORF1ab V1118I; C21846T, spike T95I; deletion 21986-21991, Spike 142/143 GV; T23542C, synonymous. To ascertain that these mutations were not assembly artifacts, we confirmed their presence in the raw sequencing data (Figure 3).

Figure 3. Screenshot illustrating reads of sample LBI215 mapped against the SARS-CoV-2 reference genome. Highlighted SNPs emphasize they factually occur in raw sequencing data and that mutations found are not assembly artifacts. SNPs shown correspond to non-synonymous mutations in the spike gene (G23012C: Spike E484Q, A23064C: Spike N501T).
Even though we found only two samples belonging to this new variant (estimated frequency: 0,02), it harbors a myriad of mutations - including four in the spike protein - possibly associated with functional effects that could lead to epidemiological consequences. These samples were epidemiologically unconnected, being collected in different laboratories from completely unrelated individuals in different regions of the MRBH, reinforcing this lineage circulation. Both genomes LBI215 and LB218 harbor new non-synonymous mutations on the critical 484 and 501 positions in the spike protein, already described increasing virus transmission and immune escape (Figure 2). The plethora of new mutations and our phylogenetic analysis suggest that this putative novel VOI was derived from lineage B.1.1.28 and should be designated as P.4. However, the PANGO nomenclature system defines that at least five genome sequences with coverage greater than 95% must be reported for a lineage to be formally classified. We are increasing the sampling of positive COVID-19 patients at the same regions to possibly identify new genomes with this signature to define this new lineage.
Conclusions
The presented results emphasize the co-circulation of VOCs B.1.1.7, P.1 and VOI P.2 in elevated frequency and a novel variant defined by multiple new mutations at positions with biological significance. The co-circulation of these variants, harboring mutations characterized by enhanced transmissibility and immune escape, is consistent with the recent increase in the number of cases and deaths observed in MRBH and other regions in Brazil (https://covid19br.github.io/) (1). B.1.1.7 lineage has been associated with increasing rates of mortality of 60% in the UK and shares the spike N501Y mutation with P.1 (1, 10).
Ongoing molecular modeling and docking experiments in spike proteins harboring the new N501T and E484Q mutations identified here are currently being conducted to predict biological impact of these mutations in virus entry, transmission and immune escape. Moreover, the recurrent emergence of diverse VOC/VOIs sharing multiple mutations at the same genome position emphasizes a pattern of consistent evolutionary convergence in SARS-CoV-2 (21). It is possible that the continuous variant emergence is related to increased virus transmission rates.
We intend to expand our sampling size and genetically characterize recent samples from the MRBH to document changes in the relative frequencies of circulating VOC/VOIs. In addition, the biological relevance of the new spike mutations described here (N501T and E484Q) will be evaluated by molecular docking with ACE2 cellular receptor and virus entry on cellular models. The novel lineage herein reported requires continued monitorization by genomic surveillance programs to better evaluate its circulation in other regions in Brazil.
References
1 - Faria NR, Claro IM, Cândido D, et al. Genomics and epidemiology of a novel SARS-CoV-2 lineage in Manaus, Brazil. CADDE GitHub page. Date accessed: April 4, 2021. https://github.com/CADDE-CENTRE/Novel-SARS-CoV-2-P1-Lineage-in-Brazil/blob/main/manuscript/FINAL_P1_MANUSCRIPT_25-02-2021_combined.pdf
2 - Voloch CM, Francisco Jr R da S, Almeida LGP de, et al. Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. Journal of Virology. 2021. https://doi.org/10.1128/JVI.00119-21
3 - Resende PR, Gräf T, Paixão ACD, et al. A potential SARS-CoV-2 variant of interest (VOI) harboring mutation E484K in the Spike protein was identified within lineage B.1.1.33 circulating in Brazil. Virological. Date accessed: April 4, 2021. https://pando.tools/t/a-potential-sars-cov-2-variant-of-interest-voi-harboring-mutation-e484k-in-the-spike-protein-was-identified-within-lineage-b-1-1-33-circulating-in-brazil/645
4 –Resende PR, Gräf T, Lima Neto LG et al. Identification of a new B.1.1.33 SARS-CoV-2 Variant of Interest (VOI) circulating in Brazil with mutation E484K and multiple deletions in the amino (N)-terminal domain of the Spike protein. Virological. Date accessed April 7 2021. https://pando.tools/t/identification-of-a-new-b-1-1-33-sars-cov-2-variant-of-interest-voi-circulating-in-brazil-with-mutation-e484k-and-multiple-deletions-in-the-amino-n-terminal-domain-of-the-spike-protein/675
5 – Slavov SN, Patané JSL, Bezerra RS, et al. Genomic monitoring unveil the early detection of the SARS-CoV-2 B.1.351 lineage (20H/501Y.V2) in Brazil. medRxiv. 2021. Genomic monitoring unveil the early detection of the SARS-CoV-2 B.1.351 lineage (20H/501Y.V2) in Brazil | medRxiv
6 - Francisco Jr R da S, Benites LF, Lamarca AP, et al. Pervasive transmission of E484K and emergence of VUI-NP13L with evidence of SARS-CoV-2 co-infection events by two different lineages in Rio Grande do Sul, Brazil. Virus Res. 2021; 296:1-7. https://doi.org/10.1016/j.virusres.2021.198345
7 - Volz E, Mishra S, Chand M, et al. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. medRxiv. 2021. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data
8 - Tegally H, Wilkinson E, Giovanetti M, et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature. 2021. Detection of a SARS-CoV-2 variant of concern in South Africa | Nature
9 - Cele S, Gazy I, Jackson L. et al. Escape of SARS-CoV-2 501Y.V2 from neutralization by convalescent plasma. Nature. 2021. https://doi.org/10.1038/s41586-021-03471
10 – Davies NG, Jarvis CI, CMMID COVID-19 Working Group, et al. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature. 2021. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7 | Nature
11 - de Souza Santos AA, Candido DdS, de Souza, WM, et al. Dataset on SARS-CoV-2 non-pharmaceutical interventions in Brazilian municipalities. Sci Data 2021; 8(73). Dataset on SARS-CoV-2 non-pharmaceutical interventions in Brazilian municipalities | Scientific Data
12 - de Souza WM, Buss LF, Candido DdS, et al. Epidemiological and clinical characteristics of the COVID-19 epidemic in Brazil. Nat Hum Behav 2020; 4:856–865. Epidemiological and clinical characteristics of the COVID-19 epidemic in Brazil | Nature Human Behaviour
13 – Lu X, Wang L, Sakthivel SK, et al. US CDC Real-Time Reverse Transcription PCR Panel for Detection of Severe Acute Respiratory Syndrome Coronavirus 2. Emerging Infectious Diseases, 2020; 26(8):1654-1665. US CDC Real-Time Reverse Transcription PCR Panel for Detection of Severe Acute Respiratory Syndrome Coronavirus 2 - Volume 26, Number 8—August 2020 - Emerging Infectious Diseases journal - CDC.
14 - Moreira FRR, Menezes DB, Zauli DAG, et al. Emergence and spread of SARS-CoV-2 lineage B.1.1.7 in Brazil. Virological. Date accessed: April 4, 2021. https://pando.tools/t/emergence-and-spread-of-sars-cov-2-lineage-b-1-1-7-in-brazil/638
15 - Rambaut A, Holmes EC, O’Toole Á. et al. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat Microbiol 2020; 5:1403–1407. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology | Nature Microbiology
16 - Davies NG, Abbott S, Barnard RC, et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science. 2021. https://doi.org/10.1126/science.abg3055
17 - Minh BQ, Schmidt HA, Chernomor O, et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol. 2020; 37(5):1530–4. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era | Molecular Biology and Evolution | Oxford Academic
18 - Tavaré S. Some probabilistic and statistical problems in the analysis of DNA sequences. Vol. 17, American Mathematical Society: Lectures on Mathematics in the Life Sciences. 1986. p. 57–86.
19 - Yang Z. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: Approximate methods. J Mol Evol. 1994; 39(3):306–14. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: Approximate methods - Journal of Molecular Evolution
20 - Guindon S, Dufayard JF, Lefort V, et al. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3. Syst. Bio. 2010; 59(3):307–21. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0 | Systematic Biology | Oxford Academic
21 - Martin D, Weaver S, Tegally H, et al. Virological. 2021. Date accessed: April 7, 2021. https://pando.tools/t/the-emergence-and-ongoing-convergent-evolution-of-the-n501y-lineages-coincided-with-a-major-global-shift-in-the-sars-cov-2-selective-landscape/618
Data sharing
All consensus genome sequences characterized in this study have been deposited on GISAID (IDs: EPI_ISL_1494960 to 1495042, EPI_ISL_1497548, EPI_ISL_1497549). Additionally, mapped reads (bam files) for samples 215 and 218, which belong to a putative novel lineage, have been uploaded to the project GitHub page.
Acknowledgments
We would like to thank all authors who submitted their data to GISAID and the EpiCoV curation team for their work. A full list of acknowledgments is available here.
Funding
We acknowledge support from the Rede Corona-ômica BR MCTI/FINEP affiliated to 116 RedeVírus/MCTI (FINEP 01.20.0029.000462/20, CNPq 404096/2020-4). This project was also supported by CNPq (R.S.A.: 312688/2017-2 and 439119/2018-9; R.P.S.: 310627/2018-4), MEC/CAPES 118 (14/2020 - 23072.211119/2020-10), FINEP (0494/20 01.20.0026.00), UFMG-NB3, FINEP nº 1139/20 (RSA), FAPEMIG (R.P.S.: APQ-00475-20) and FAPERJ (C.M.V: 26/010.002278/2019) (R.S.A 202.922/2018) and by CADDE/FAPESP (MR/S0195/1 and FAPESP 120 18/14389-0) (NRF). WMS is supported by FAPESP #2017/13981-0, 2019/24251-9 and CNPq #408338/2018-0.