Identification of SARS-CoV-2 P.1-related lineages in Brazil provides new insights about the mechanisms of emergence of Variants of Concern
Tiago Gräf 1A, Gonzalo Bello 2A, Taina Moreira Martins Venas 3, Elisa Cavalcante Pereira 3, Anna Carolina Dias Paixão 3; Luciana Reis Appolinario 3; Renata Serrano Lopes 3; Ana Carolina da Fonseca Mendonça 3; Alice Sampaio Barreto da Rocha 3; Fernando Couto Motta 3, Tatiana Schäffer Gregianini 4, Richard Salvato 4, Sandra Bianchini Fernandes 5, Darcita Buerger Rovaris 5, Andrea Cony Cavalcanti 6, Anderson Brandão Leite 7, Irina Riediger 8, Maria do Carmo Debur 8, André Felipe Leal Bernardes 9, Rodrigo Ribeiro-Rodrigues10, Beatriz Grinsztejn11, Filipe Zimmer Dezordi12,13, Gabriel Luz Wallau 12,13B, Felipe Gomes Naveca 14B, Edson Delatorre 15B, Marilda Mendonça Siqueira 3B, and Paola Cristina Resende 3B
on behalf of Fiocruz COVID-19 Genomic Surveillance Network.
A These authors contributed equally.
B These authors share the senior authorship.
1 Plataforma de Vigilância Molecular, Instituto Gonçalo Moniz, Fiocruz, Salvador, Bahia, Brazil.
2 Laboratório de AIDS e Imunologia Molecular, Instituto Oswaldo Cruz, Fiocruz, Rio de Janeiro, Brazil.
3 Laboratório de Vírus Respiratórios e do Sarampo (LVRS), Instituto Oswaldo Cruz, Fiocruz, Rio de Janeiro, Brazil.
4 Laboratório Central de Saúde Pública do Estado do Rio Grande do Sul (LACEN-RS), Brazil.
5 Laboratório Central de Saúde Pública do Estado de Santa Catarina (LACEN-SC), Brazil.
6 Laboratório Central de Saúde Pública do Estado do Rio de Janeiro (LACEN-RJ), Brazil.
7 Laboratório Central de Saúde Pública do Estado de Alagoas (LACEN-AL), Brazil.
8 Laboratório Central de Saúde Pública do Estado do Paraná (LACEN-PR), Brazil.
9 Laboratório Central de Saúde Pública do Estado de Minas Gerais (LACEN-MG), Brazil.
10 Laboratório Central de Saúde Pública do Estado do Espírito Santo (LACEN-ES), Brazil.
11 Instituto Nacional de Infectologia (INI), Fiocruz, Rio de Janeiro, Brazil.
12 Departamento de Entomologia, Instituto Aggeu Magalhães, Fiocruz, Recife, Pernambuco, Brazil.
13 Núcleo de Bioinformática (NBI), Instituto Aggeu Magalhães Fiocruz, Recife, Pernambuco, Brazil.
14 Laboratório de Ecologia de Doenças Transmissíveis na Amazônia (EDTA), Instituto Leônidas e Maria Deane, FIOCRUZ, Manaus, Amazonas, Brazil.
15 Departamento de Biologia. Centro de Ciências Exatas, Naturais e da Saúde, Universidade Federal do Espírito Santo, Alegre, Brazil.
Abstract
One of the most remarkable features of the SARS-CoV-2 Variants of Concern (VOC) is the unusually large number of mutations they carry, but the precise factors that drove the emergence of such variants since the second half of 2020 are not fully resolved. In this study we described a new SARS-CoV-2 lineage provisionally designated as P.1-like-II that, as well as the previously described lineage P.1-like-I, shares several lineage-defining mutations with the VOC P.1 circulating in Brazil. Reconstructions of P.1 ancestor sequences demonstrate that the full constellation of mutations that define the VOC P.1 did not accumulate within a single long-term infected individual, but were acquired at sequential steps during multiple rounds of infections. Our analyses further estimate that a P.1-ancestor carrying half of the P.1-lineage-defining mutations, including those at the receptor-binding domain of the Spike protein, has been probably circulating cryptically in the Amazonas state since August 2020. This evolutionary pattern is consistent with the hypothesis that partial human population immunity acquired from natural SARS-CoV-2 infections during the first half of 2020 might have been the major driving force behind natural selection that allowed the emergence and worldwide spread of VOCs.
1. Introduction
The emergence of the SARS-CoV-2 variant of concern (VOC) P.1 in the Brazilian Amazonas state around November 2020 [1, 2] and its rapid dissemination to other regions was associated with a major COVID-19 epidemic wave that collapsed the Brazilian public health system during early 2021. The lineage P.1, as the other VOCs described, harbors a large number of lineage-defining mutations, including: 10 non-synonymous substitutions in the Spike (S) protein (L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, H655Y, T1027I), five non-synonymous mutations distributed in the NSP3 (S370L and K977Q), NSP13 (E341D), NS8 (E92K) and N (P80R) proteins, one deletion in the NSP6 (S106del, G107del, F108del) and a four-nucleotide insertion at ORF8/N intergenic region (ins28263) [1, 2].
The most accepted hypothesis to explain such a high number of lineage-defining mutations is that VOCs result from selective pressures and adaptation of the virus during prolonged infections in immunosuppressed individuals [1, 2]. However, the analysis of a large number of SARS-CoV-2 viruses in Amazonas revealed four P.1-like sequences, mostly sampled in Manaus, that branched as a sister monophyletic clade with respect to lineage P.1 [1, 3]. The P.1-like clade also accumulated an unusually high number of genetic changes relative to the parental B.1.1.28 lineage, including several P.1 lineage-defining mutations in the S (L18F, P26S, D138Y, K417T, E484K, N501Y), NSP3 (K977Q) and N (P80R) proteins and also unique mutations in the NSP2 (K456R), NSP3 (T1189I), NSP6 (V149A), NSP13 (S74L), S (ins214 and D1139H) and NS8 (K2stop) proteins. This finding suggests that P.1 lineage-defining mutations did not accumulate in a unique long-term infection event, but were acquired at sequential steps during the evolution of lineage B.1.1.28 in the Amazonas state.
In this study, we described a second P.1-related virus variant that is spreading in several states from the different Brazilian regions and harbors 15 P.1 lineage-defining mutations and six unique mutations. The description of this new P.1-related variant allowed us to trace with more precision the evolutionary steps that resulted in the emergence of the VOC P.1 and confirms that some of the P.1 lineage-defining mutations were sequentially fixed over several months during the second half of 2020. Our analyses also revealed that despite sharing crucial mutations in the RBD of the S protein, the P.1-like variants displayed a much less efficient epidemic spread in Brazil compared with the VOC P.1.
2. Materials and Methods
Our genomic survey of SARS-CoV-2 positive samples sequenced by the Fiocruz COVID-19 Genomic Surveillance Network between 12th March 2020 and 31st March 2021 identified 44 sequences (EPI_ISL_2038926 to EPI_ISL_2038968, EPI_ISL_2102018 and EPI_ISL_2102063, Supplementary Table 1.pdf (586.3 KB)) with several overlapping mutations with the lineage P.1. The SARS-CoV-2 genomes were recovered using Illumina sequencing protocols as previously described [4, 5]. The FASTQ reads obtained were imported into the CLC Genomics Workbench version 20.0.4 (Qiagen A/S, Denmark), trimmed, and mapped against the reference sequence EPI_ISL_402124 available in EpiCoV database in the GISAID (https://www.gisaid.org/ 4). The alignment was refined using the InDels and Structural Variants module. This study was approved by the FIOCRUZ-IOC (68118417.6.0000.5248 and CAAE 32333120.4.0000.5190) and the Amazonas State University Ethics Committee (CAAE: 25430719.6.0000.5016) and the Brazilian Ministry of the Environment (MMA) A1767C3.
SARS-CoV-2 P.1-related sequences here obtained were aligned with high quality (<5% of N) and complete (>29 kb) sequences that were available in EpiCoV database in the GISAID (https://www.gisaid.org/) on March 31st, 2021 and belongs to three different clades: 1) B.1.1.28 sequences from Amazonas state, 2) P.1 sequences, and 3) previously described P.1-like sequences [1, 3]. This dataset was then aligned using MAFFT v7.475 [6] and subjected to maximum likelihood (ML) phylogenetic analysis using IQ-TREE v2.1.2 [7] under the GTR+F+R4 nucleotide substitution model, as selected by the ModelFinder application [8]. Branch support was assessed by the approximate likelihood-ratio test based on the Shimodaira–Hasegawa procedure (SHaLRT) with 1000 replicates. The sequence of ancestral nodes was reconstructed using Time-tree [9] and their mutational profile investigated using the Nextclade tool (https://clades.nextstrain.org). The temporal signal was assessed by the regression analysis of the root-to-tip genetic distance against sampling dates using the program TempEst [10].
A time-scaled phylogenetic tree of the B.1.1.28 Amazonian diversity plus a subsampling of P.1 genomes and P.1-related sequences was reconstructed using the Bayesian Markov Chain Monte Carlo (MCMC) approach implemented in BEAST 1.10.4 [11]. A Bayesian tree was reconstructed using the GTR+F+G4 nucleotide substitution model, the Bayesian skyline (BSKL) coalescent model [12], and both strict and random local molecular clock models [13] with a uniform substitution rate prior (8 x 10–4 – 10 x 10–4 substitutions/site/year). Ancestral sampling locations were inferred using a reversible discrete phylogeographic model [14] where transitions between Brazilian states were estimated in a continuous-time Markov chain (CTMC) rate reference prior. Convergence (effective sample size > 200) in parameter estimates was assessed using TRACER v1.7 [15]. The maximum clade credibility (MCC) tree was summarized with TreeAnnotator v1.10.4. ML and MCC trees were visualized using FigTree v1.4.4 (FigTree).
3. Results and Discussion
Mutation profile analysis of SARS-CoV-2 positive samples detected at different Brazilian states between 12th March 2020 and 31st March 2021 revealed 44 sequences ( Supplementary Table 1.pdf (586.3 KB)) that harbor 15 out of 22 P.1 lineage-defining mutations, including the three mutations of concern at the receptor-binding domain (RBD) of the S protein (K417T, E484K and N501Y), deletion in the NSP6 (S106del, G107del, F108del) and the four-nucleotide insertion at ORF8/N intergenic region (ins28263). These P.1-related sequences, here designated as P.1-like-II, lack some of the P.1 lineage-defining mutations at ORF1ab (C2749T, C12778T and C13860T), NSP13 (E341D), S (T20N) and NS8 (E92K) and further displayed six unique substitutions at ORF1ab (C8905T, C16954T, and A20931G), NSP4 (D217H), E/M intergenic region (A26492T) and N (P383L). The P.1-like-II sequences also share nine P.1 lineage-defining mutations with the previously characterized P.1-like clade (now designated as P.1-like-I).
ML phylogenetic analysis revealed that new P.1-like-II sequences branched in a highly supported (SH-aLRT = 96.6%) monophyletic clade together with seven sequences retrieved from the EpiCoV database (https://www.gisaid.org/) that displayed the P.1-like-II mutation profile, but were currently classified as P.1 in the EpiCoV database (Figure 1a). The clades P.1-like-I and P.1-like-II are not nested within the diversity of the VOC P.1 and then should be designated as new “P.n” PANGO lineages (request for lineage designation submitted on 10th May, 2021 New SARS-CoV-2 P.1-related lineages proposal - P.1-like-I and P.1-like-II in Brazil · Issue #77 · cov-lineages/pango-designation · GitHub). The P.1-like-II genomes were sampled at nine different Brazilian states, mainly from the South and Southeast regions (Figure 1b), the oldest one was detected in the Rio de Janeiro state on 19th January 2021 [16] and the most recent one was identified in this study in the Amazonas state on 25th March 2021. In contrast to the SARS-CoV-2 lineages P.1 and P.1-like-I, we found almost no evidence of dissemination of lineage P.1-like-II within the Amazonian region. The Brazilian state that comprises most P.1-like-II sequences identified so far was Santa Catarina (59%), followed by Rio de Janeiro (10%), Rio Grande do Sul (8%) and Sao Paulo (8%).
Analysis of the temporal structure revealed that P.1 and P.1-like clades accumulated a higher number of mutations than other B.1.1.28 sequences (Figure 1c). Reconstruction of sequences at ancestral nodes provides a clear picture of the evolutionary steps that resulted in the different P.1 and P.1-related lineages (Figure 2). Three mutations were fixed in the basal B.1.1.28 Amazonian clade 28-AM-II [1], from which all P.1 and P.1-related viruses evolved. Nine mutations were fixed in the following evolutionary step that gave origin to the most recent common ancestor (MRCA) of all P.1 and P.1-related viruses (designated as P.1MRCA1). Six out of the nine (67%) mutations in P.1MRCA1 were in the S protein, including the three mutations of concern in the RBD. In sharp contrast, out of 32 mutations fixed in the different branches that originated from P.1MRCA1 only seven (22%) were located in the S gene. It is also interesting to note that the total number of lineage-defining mutations accumulated by P.1 (n = 12), P.1-like-I (n = 14) and P.1-like-II (n = 12) since their divergence from P.1MRCA1 was almost the same, thus suggesting that those viral lineages evolve at similar rate over time.
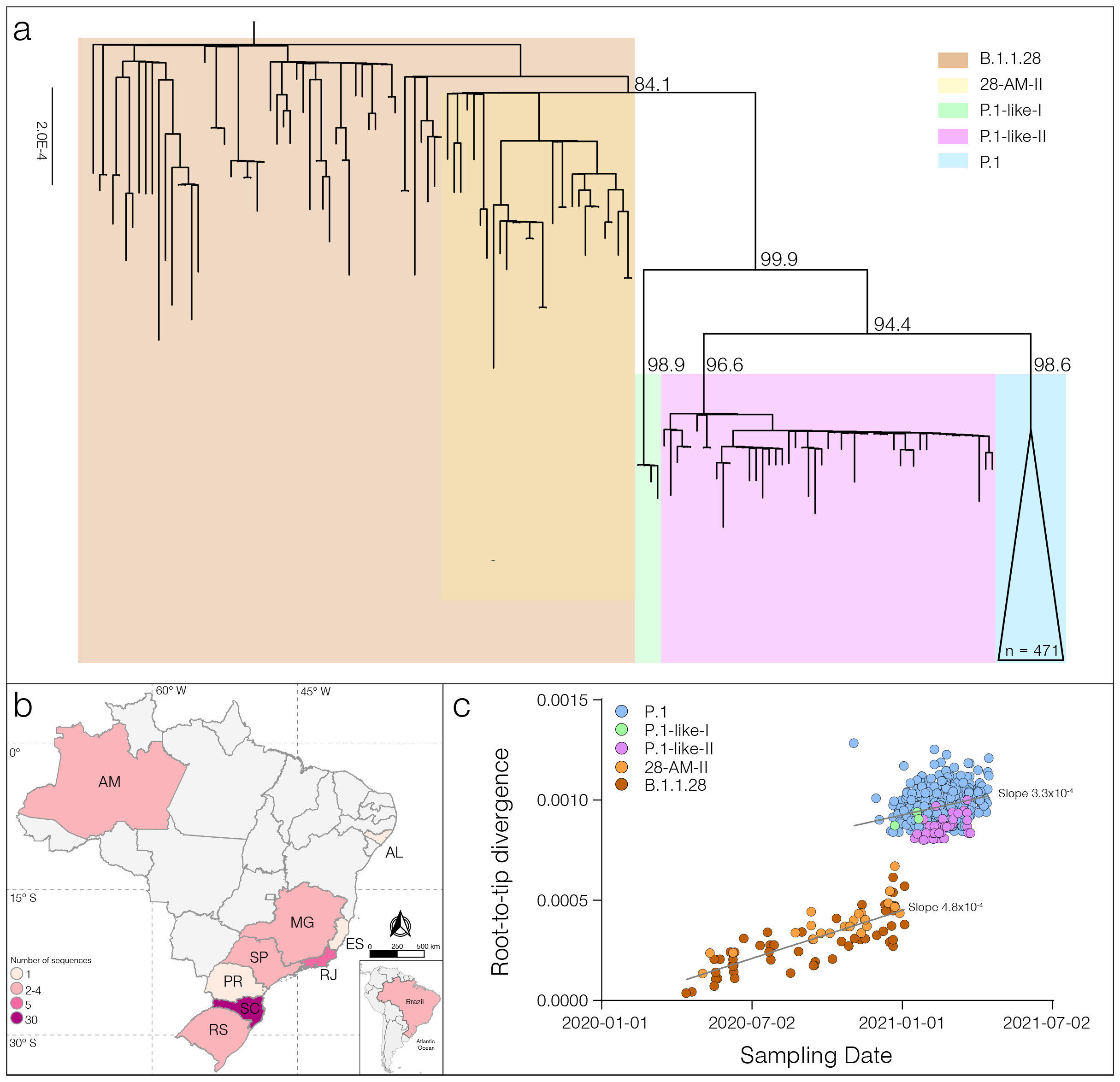
Figure 1. Genetic diversity and distribution of the B.1.1.28, P.1 and P.1-like lineages in Brazil. a) Maximum likelihood (ML) phylogenetic tree of the B.1.1.28, P.1, and P.1-like lineages identified in Brazil. Each lineage was highlighted with colored boxes as indicated in the legend. The aLRT support values are indicated in key branches, and branch lengths are drawn to scale with the lateral bar indicating nucleotide substitutions per site. b) Geographic distribution and frequency of the P.1-like-II lineage identified in Brazil. Brazilian states’ names follow the ISO 3166-2 standard. Color’s gradient represents the number of sequences identified in this study, following the legend. c) Correlation between the sampling date of B.1.1.28, P.1, P.1-like-I, and P.1-like-II and their genetic distance from the ML phylogenetic tree’s root. Each lineage was colored following the legend. The slope of each regression is indicated.
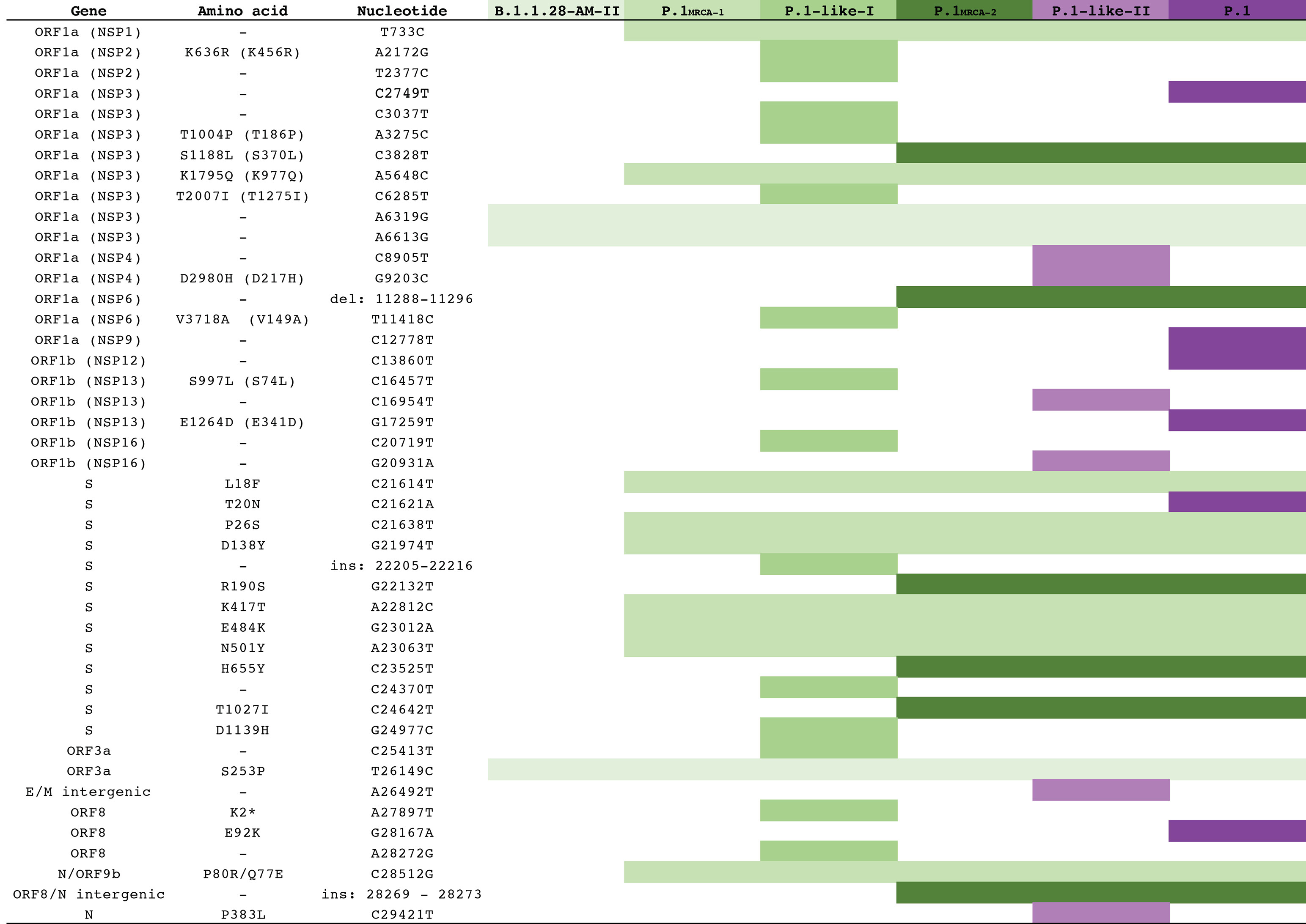
Figure 2. Evolutionary steps associated with the emergence of the P.1 and P.1-related lineages. Each line represents a mutation that emerged during the diversification of the B.1.1.28 lineage in Brazil originating the P.1, P.1-like-I and P.1-like-II.
Bayesian phylogeographic analysis was next conducted combining all B.1.1.28 sequences from Amazonas (including clade 28-AM-II), early P.1 viruses sampled in December 2020 and all P.1-related sequences. This analysis support that most ancestors of P.1 and P.1-related viruses probably arose in the state of Amazonas (Posterior State Probability [PSP] = 1), with the only exception of the P.1-like-II ancestor whose origin was traced to Amazonas (PSP = 0.40) and Santa Catarina (PSP = 0.31) with similar probability (Figure 3).
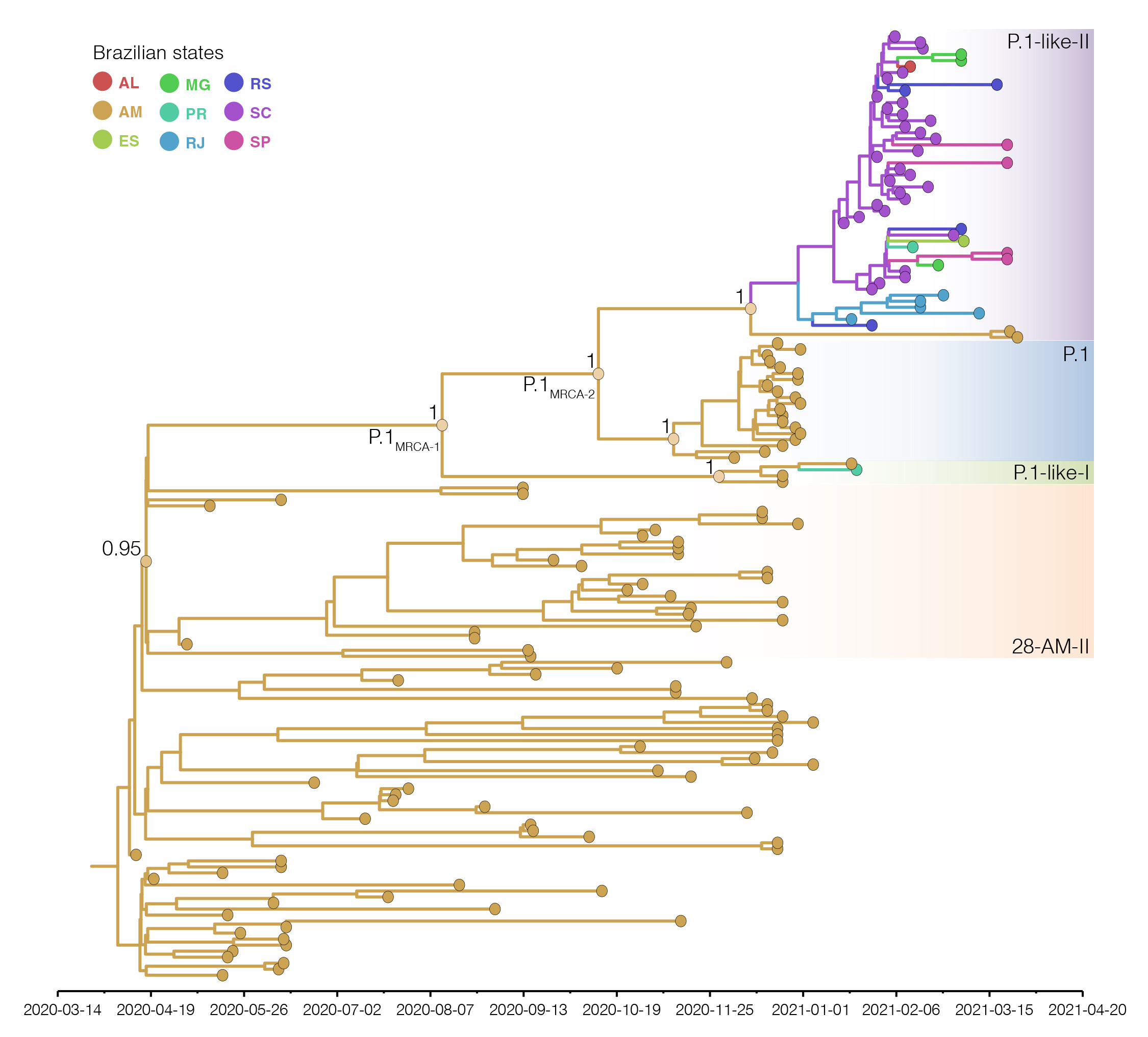
Figure 3. Bayesian phylogeographic analysis of the B.1.1.28, P.1 and P.1-related lineages. Tips and branches colors indicate the Brazilian state (ISO 3166-2 standard) of sampling and the most probable inferred location of their descendent nodes, respectively, as indicated in the legend. Branch posterior probabilities are indicated in key nodes. Boxes with different colors highlight the 28-AM-II, P.1, P.1-like-I and P.1-like-II lineages. All horizontal branch lengths are time-scaled and the tree was automatically rooted under the assumption of the strict molecular clock model. Reconstructed ancestral key nodes representing the most recent common ancestor (MRCA) of each lineage and the MRCA of all P.1 and P.1-related viruses (labeled as P.1MRCA1) and the MRCA of P.1 and P.1-like-II (labeled as P.1MRCA2) are highlighted with circles.
The great uncertainty in the location of the P.1-like-II ancestor probably reflects the very low number of sequences from this clade detected in the Amazonas state so far, making it difficult to trace their origin to that Northern state. This analysis estimated that Santa Catarina was the most important hub of dissemination of lineage P.1-like-II to other Brazilian states. It is also noteworthy that P.1-like-II genomes from Rio de Janeiro formed a basal cluster in the tree, supporting local transmission of this lineage has been established in this state. The different molecular clock models used consistently traced the median time of the P.1MRCA1 to mid-August 2020, the median time of the P.1MRCA2 to late September 2020, and the emergence of lineages P.1 and P.1-related variants to around late November and late December 2020 (Table), respectively, consistent with previous estimates [1, 2].
Table. Bayesian estimates of the time of P.1 and P.1-related most recent common ancestors using two different molecular clock models.
| Ancestor | Strict clock | Random local clock |
|---|---|---|
| P.1MRCA1 | 12th Aug 2020 (05th Jul - 17th Sep) | 11th Aug 2020 (02th Jul - 16th Sep) |
| P.1MRCA2 | 30th Sep 2020 (27th Aug - 29th Oct) | 30th Sep 2020 (27th Aug - 31st Oct) |
| P.1 | 22nd Nov 2020 (6th Nov - 04th Dec) | 22nd Nov 2020 (6th Nov - 04th Dec) |
| P.1-like-I | 18th Dec 2020 (9th Dec - 23rd Dec) | 18th Dec 2020 (9th Dec - 23rd Dec) |
| P.1-like-II | 28th Dec 2020 (13th Dec - 08th Jan) | 27th Dec 2020 (12th Dec - 07th Jan) |
In contrast to the most widely accepted hypothesis that suggests that mutations in VOCs arose during long-standing SARS-CoV-2 infections in immunosuppressed individuals, our findings revealed that VOC P.1 defining mutations were acquired through multiple inter-host transmissions events. Furthermore, our time-scale estimates using both strict and random local molecular clock models suggest that the final constellation of mutations observed in lineage P.1 was not acquired in a short time interval, but was an evolutionary process that probably occurred over several months of communitarian transmission. Although this pattern does not exclude the possibility that at least a subset of P.1 mutations could have originated in immunosuppressed individuals, sequential infection of such patients is very unlikely. We propose that such mutations have been naturally selected during acute reinfection of partially protected immunocompetent individuals. According to this hypothesis, the partial immunity that human populations acquired through natural SARS-CoV-2 infections during early 2020 was a major selective force that drove the sequential emergence of mutations of concern in the second half of 2020. This model is consistent with a recent study that revealed a major change in selective pressures acting on SARS-CoV-2 variants circulating worldwide after October 2020, coinciding with the simultaneous expansion of different VOCs with convergent mutations in Europe, Africa and South America [17].
4. Conclusions
Our genomic surveillance identified a new P.1-realted clade derived from the B.1.1.28 Amazonian diversity that was provisionally designated as lineage P.1-like-II. It shares a common ancestor and several lineage-defining mutations with the VOC P.1, including those in the RBD of the S protein (K417T, E484K, N501Y), and is geographically dispersed in Brazil, particularly in the South and Southeast country regions. The P.1-like-II is the second P.1-related lineage identified by our group in Brazil, confirming that the full constellation of mutations that defines the VOC P.1 did not accumulate in a single individual, but during multiple inter-host transmission events. Our findings further suggest that a P.1-ancestor carrying half of the P.1-lineage-defining mutations, including those at the RBD of the S protein, circulates cryptically in the Amazonas state for several months before the emergence of P.1 and P.1-related lineages. Of note, although the P.1-ancestor and the contemporaneous P.1-related lineages displayed the key mutations of concern in the S protein; none of them was so efficiently spread as VOC P.1. This suggests that factors other than viral mutations, most likely related to human behavior, might have played a role in the remarkable dissemination of the VOC P.1 in the Amazonas state and throughout Brazil afterwards.
Acknowledgments
The authors wish to thank all the health care workers and scientists who have worked hard to deal with this pandemic threat, the GISAID team, and all the EpiCoV database′s submitters, GISAID acknowledgment table containing sequences used in this study is available in Supplementary Table S2 GISAID_Acknowledgement_Table_S2.pdf (102.9 KB). We also appreciate the support of the Fiocruz COVID-19 Genomic Surveillance Network (http://www.genomahcov.fiocruz.br/; accessed on May 2021) members, the Respiratory Viruses Genomic Surveillance. General Coordination of the Laboratory Network (CGLab), Brazilian Ministry of Health (MoH), Brazilian States Central Laboratories (LACEN).
Funding
Financial support was provided by Fundação de Amparo à Pesquisa do Estado do Amazonas (FAPEAM) (PCTI-EmergeSaude/AM call 005/2020 and Rede Genômica de Vigilancia em Saúde-REGESAM); Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (grant 402457/2020–0); CNPq/Ministério da Ciência, Tecnologia, Inovações e Comunicações/Ministério da Saúde (MS/FNDCT/SCTIE/Decit) (grant 403276/2020-9); Inova Fiocruz/Fundação Oswaldo Cruz (Grants VPPCB-007-FIO-18–2–30 and VPPCB-005-FIO-20–2–87), INCT-FCx (465259/2014–6) and Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) (26/210.196/2020). F.G.N, G.L.W, G.B and M.M.S are supported by the CNPq through their productivity research fellowships (306146/2017–7, 303902/2019–1, 302317/2017–1 and 313403/2018-0, respectively). G.B. is also funded by FAPERJ (Grant number E-26/202.896/2018).
References
1 Naveca, F. et al. COVID-19 epidemic in the Brazilian state of Amazonas was driven by long-term persistence of endemic SARS-CoV-2 lineages and the recent emergence of the new Variant of Concern P.1. Research Square, doi:10.21203/rs.3.rs-275494/v1 (2021).
2 Faria, N. R. et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science, doi:10.1126/science.abh2644 (2021).
3 Resende, P. C. et al. The ongoing evolution of variants of concern and interest of SARS-CoV-2 in Brazil revealed by convergent indels in the amino (N)-terminal domain of the Spike protein. medRxiv, doi:10.1101/2021.03.19.21253946 (2021).
4 Nascimento, V. A. D. et al. Genomic and phylogenetic characterisation of an imported case of SARS-CoV-2 in Amazonas State, Brazil. Mem Inst Oswaldo Cruz 115, e200310, doi:10.1590/0074-02760200310 (2020).
5 Resende, P. C. et al. SARS-CoV-2 genomes recovered by long amplicon tiling multiplex approach using nanopore sequencing and applicable to other sequencing platforms. bioRxiv, doi:10.1101/2020.04.30.069039 (2020).
6 Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30, 772-780, doi:10.1093/molbev/mst010 (2013).
7 Minh, B. Q. et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol 37, 1530-1534, doi:10.1093/molbev/msaa015 (2020).
8 Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14, 587-589, doi:10.1038/nmeth.4285 (2017).
9 Kumar, S., Stecher, G., Suleski, M. & Hedges, S. B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol Biol Evol 34, 1812-1819, doi:10.1093/molbev/msx116 (2017).
10 Rambaut, A., Lam, T. T., Max Carvalho, L. & Pybus, O. G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol 2, vew007, doi:10.1093/ve/vew007 (2016).
11 Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol 4, vey016, doi:10.1093/ve/vey016 (2018).
12 Drummond, A. J., Rambaut, A., Shapiro, B. & Pybus, O. G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol 22, 1185-1192, doi:10.1093/molbev/msi103 (2005).
13 Ferreira, M. A. R. S., M.A. Bayesian analysis of elapsed times in continuous‐time Markov chains. The Canadian Journal of Statistics 36, 355-368, doi: https://doi.org/10.1002/cjs.5550360302 (2008).
14 Lemey, P., Rambaut, A., Drummond, A. J. & Suchard, M. A. Bayesian phylogeography finds its roots. PLoS Comput Biol 5, e1000520, doi:10.1371/journal.pcbi.1000520 (2009).
15 Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst Biol, doi:10.1093/sysbio/syy032 (2018).
16 Lamarca, A. et al. Genomic surveillance of SARS-CoV-2 tracks early interstate transmission of P.1 lineage and diversification within P.2 clade in Brazil. medRxiv doi:10.1101/2021.03.21.21253418 (2021).
17 Martin, D. P. et al. The emergence and ongoing convergent evolution of the N501Y lineages coincides with a major global shift in the SARS-CoV-2 selective landscape. medRxiv, doi:10.1101/2021.02.23.2125226 (2021).