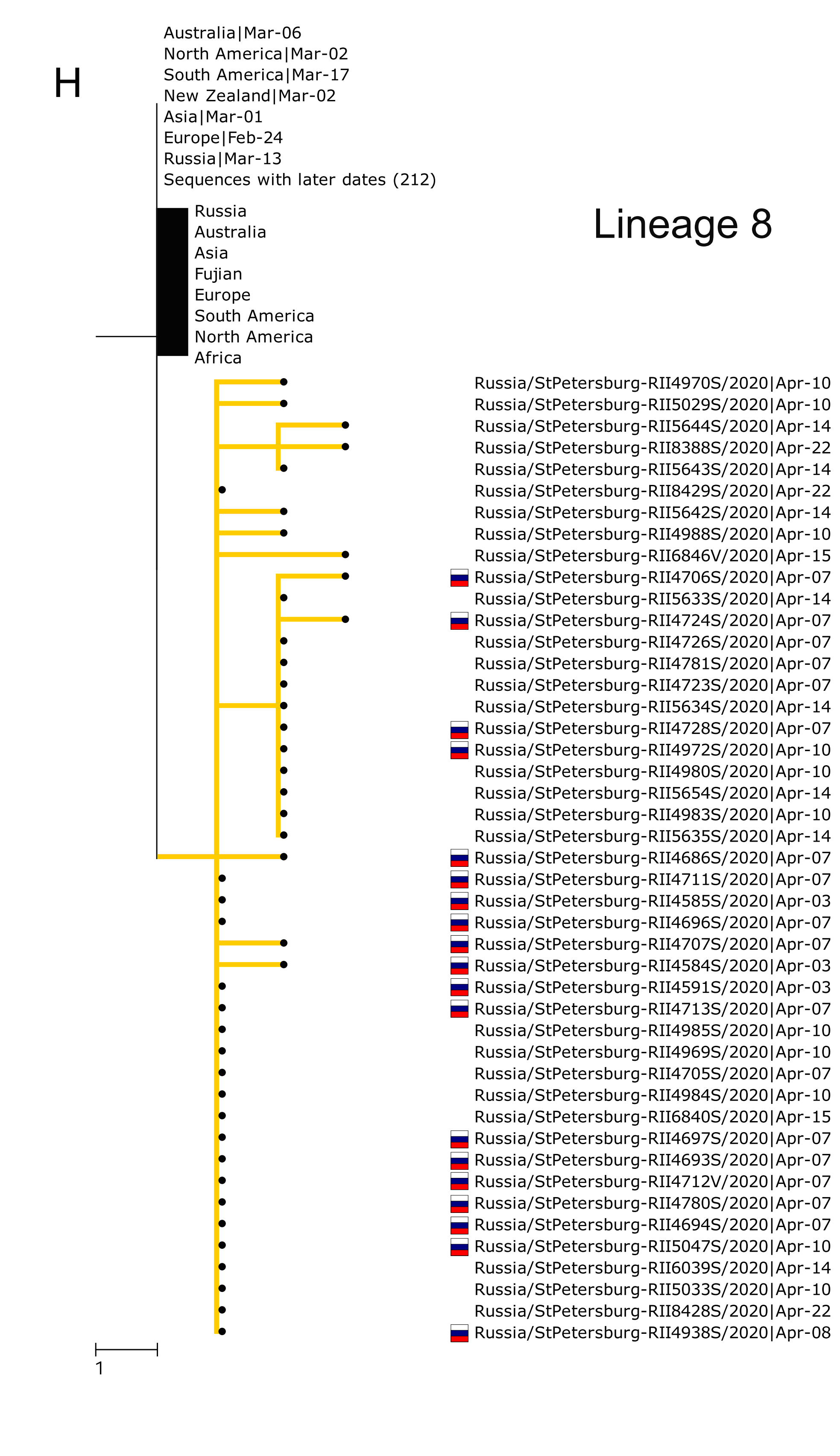
Russia ranks fourth in the number of confirmed COVID-19 cases globally. In our new preprint, we perform the first (to my knowledge) study of the genomic epidemiology of SARS-CoV-2 in Russia in March-April. This is based on 211 genomes from 25 (out of the 85) Russia’s regions.
Key findings:
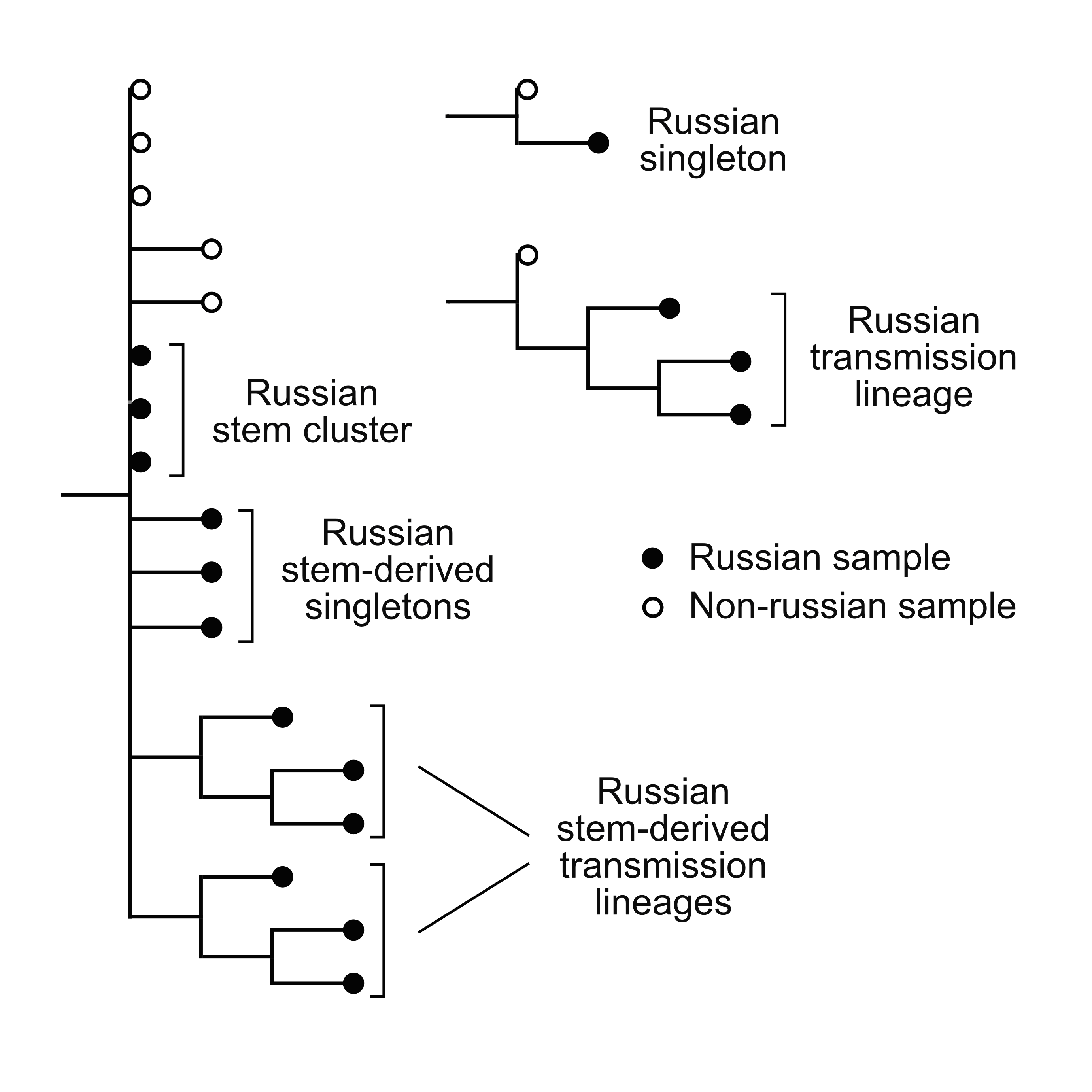
- 67 independent introductions (probably more), mostly from Europe;
- 9 Russian transmission lineages;
- phylogeographic positions of samples match direct travel data. In 9 out of 13 cases, it is consistent with the country of origin, including 3 cases when the country of origin is uniquely and correctly identified (France, Switzerland and Saudi Arabia).
- no trace of export outside Russia.
Additionally, we study a large nosocomial cluster – the Vreden hospital in Saint Petersburg. Over 700 patients and medical staff stayed there locked down for over a month; over 400 got infected. We find that the virus was introduced into the hospital up to 4 times; each introduction gave rise to an outbreak of its own, with initial Rt~4, later reduced to ~1.
https://www.medrxiv.org/content/10.1101/2020.07.14.20150979v1