Carmen Lia Murall (1)*, Eric Fournier (2)*, Jose Hector Galvez (3,4), Sarah J. Reiling (3,6), Pierre-Olivier Quirion (3,4,5), Anne-Marie Roy (3,6), Shu-Huang Chen (3,6), Paul Stretenowich (3,4), Mathieu Bourgey (3,4), Mark Lathrop (3,6), Michel Roger (1,2), Guillaume Bourque (3,4,6), Jiannis Ragoussis (3,6,7), B. Jesse Shapiro (1,3,8), Sandrine Moreira (1,2), on behalf of the CoVSeQ Consortium (9)
Affiliations:
1 Université de Montréal
2 Laboratoire de Santé Publique du Québec (LSPQ), Institut National de Santé Publique (INSPQ)
3 McGill Genome Center
4 Canadian Center for Computational Genomics
5 Calcul Québec
6 Department of Human Genetics, McGill University
7 Department of Bioengineering, McGill University
8 Department of Microbiology and Immunology, McGill University
9 https://covseq.ca/researchgroup
*equal contribution
Summary
- Québec was the hardest-hit Canadian province early in the COVID-19 pandemic with more than 62,000 positive cases as of September 1, 2020.
- We report 734 high-quality SARS-Cov-2 consensus sequences from the first month of the pandemic in the province
- Added to a global phylogeny, these sequences help refine the number of introduction events into Québec, conservatively estimated at >200 independent events by late March
- We place the inferred introduction events in the context of travel destinations and frequencies, the majority of which happened between the end of spring break and a week after the border closed
- As expected, phylogenetic analysis places the introduction events systematically earlier (during spring break) than observed from case reports and travel history (after spring break)
- Consistent with common spring break travel destinations, most of the introduced sequences were from clades common in Europe and the Americas, and only rarely from Asia
- Sequencing efforts are ongoing to track the relative transmission rates of the various SARS-CoV-2 clades introduced into Québec
Introduction
Québec is the second most populous province in Canada, and the one hardest hit in the first months of the ongoing COVID-19 pandemic. About half of its 8 million inhabitants live in the densely populated Montréal metropolitan area. The Public Health Laboratory of Québec (LSPQ) has the mandate of developing specialized diagnostic tests for the province and to respond to emergency situations. A qPCR diagnostic test targeting SARS-CoV-2 E and N genes was developed when the first cases were reported in China and Europe (LeBlanc et al. 2020). The first case of COVID-19 in Québec was detected on February 25. Shortly after, Québec was the first large Canadian province to start its spring break (February 29 to March 9, Fig. 1). It is believed that these early dates had a major impact on the epidemic. Upon returning from spring break, the criteria for getting tested and for isolation or quarantine by the Public Health authorities included symptoms of cough or fever and/or contact with infected people. The number of cases increased exponentially during March (Fig. 2, Québec COVID-19 data). On March 13th, a public health emergency was declared, and schools, and daycares were closed on March 16th as containment/lockdown began in earnest. The closing of the Canadian border was announced March 16th and officially closed the night of the 17th. On March 18th, the diagnostic test was decentralized from the LSPQ to the supra-regional hospital laboratories throughout the province. On March 20th, Québec reached the threshold of 100 cases per day. As of September 1st, Québec has been the hardest-hit Canadian province (Table 1), and suffered some of the worst death rates in the world (~ 68 per 100,000).
To better understand the early introduction and transmission events of SARS-CoV-2 in Québec, we sequenced and analyzed 734 high-quality consensus sequences obtained between mid-February and April 1, 2020. We placed these in the context of 21,935 sequences from elsewhere in Canada and internationally, including all available in GISAID up to April 1st. We searched for the geographical origins of the various introductions by comparing the results of epidemiological travel-history data with phylogenetic inference.
Fig. 1. Timeline of COVID-19 epidemic events in Québec up to April 1, 2020.
For more details of control measures and case counts: Ligne du temps COVID-19 au Québec | Institut national de santé publique du Québec

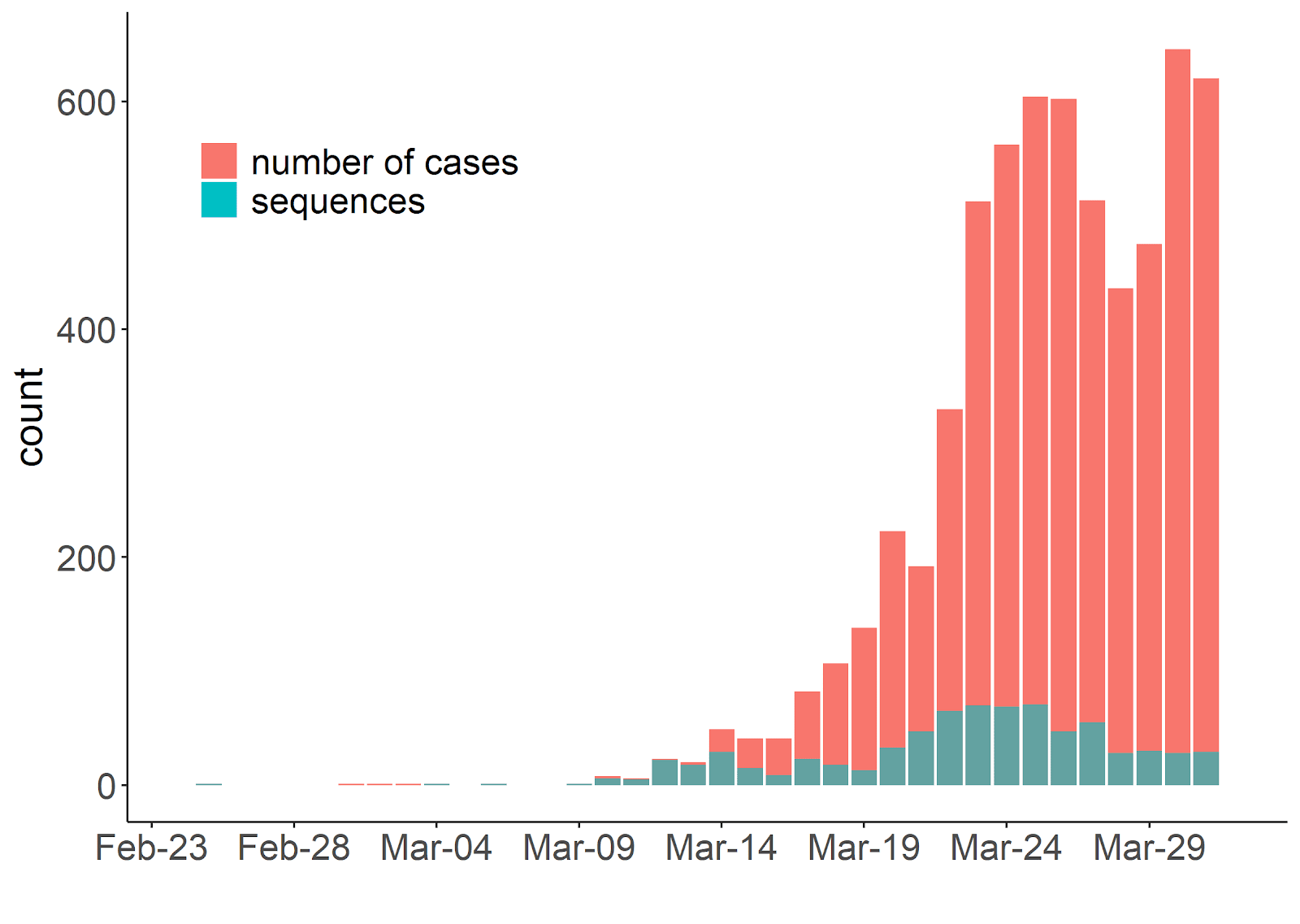
Fig. 2. Comparison of confirmed cases (red) reported by public health authorities and high-quality sequences used in this study (blue) distributed by collection date.
March 15 April 1 Number of deaths (Québec total) 0 97 Long-term care facilities 0 59 Seniors’ residence 0 14 Hospitalizations (Québec total) 2 365 Intensive Care Unit 0 96 Number of tests (Québec total) 3079 69146 Number of cases in Québec 154 5389 Total number of cases in Canada 253 9613 Table 1. Statistics of the early phase of Québec’s COVID-19 epidemic.
Sources:
Données COVID-19 au Québec | Institut national de santé publique du Québec, accessed August 19, 2020. https://www.canada.ca/en/public-health/services/diseases/2019-novel-coronavirus-infection.html accessed August 19, 2020.
Note that there is a short delay between detection at the provincial level and reporting to federal authorities.
Results
1. Sampling and sequencing
In April 2020, we assembled the Coronavirus Sequencing in Québec (CoVSeQ) consortium (https://covseq.ca/) to sequence SARS-CoV-2 from Québec cases. The CoVSeQ consortium is part of the Canadian COVID Genomic Network (CanCOGeN), a pan-Canadian cross-agency network for large-scale SARS-CoV-2 and human host sequencing (CanCOGeN - GenomeCanada). Here we present the first release of sequences, including 734 high-quality consensus sequences, from the Québec component of this collaboration (available here: CoVSeQ, https://covseq.ca/data-info ). Our sampling effort covers most of the reported cases in Québec up to March 16th (Fig. 2). We are currently in the process of sequencing more samples from late March through April to better capture the growth phase of the epidemic. Sequences of SARS-CoV-2 viral genomes were obtained by targeted amplification from clinical nasopharyngeal swabs specimens followed by sequencing on Nanopore (n=207), Illumina (n=416) or MGI (n=111) platforms. Only sequences passing our quality criteria (less than 5% undetermined bases, “Ns”) were considered for further phylogenetic analyses (Methods).
2. Estimating the number of SARS-Cov-2 introduction events into Québec
To determine how genome sequencing and phylogenetic analysis can complement and refine the identification of introduction events of SARS-CoV-2 into Québec, we compared travel history provided by patients in the laboratory requisition form with the phylogenetic inference of introductions. Of the 734 high-quality consensus sequences analyzed here, 330 were from COVID-19 cases that had reported recent travel history in our dataset (Fig. 3A), likely reflecting a tendency for introduction events during this time period, combined with the recommendation that people with a recent travel history be tested. Travel-history data suggests that most importations came from Europe (n = 108, 32.7% with the most from France, n = 40, 12.1%), the Caribbean and Latin America (n = 102, 30.9%) and the USA (n = 79, 23.9%). Few introductions came from Asia (n = 4, 1.2%) and none from China. Similar to Pybus et al. (2020), we used ancestral state reconstruction (ASR) on a global context tree, to identify a total of 367 introductions by finding non-Québec to Québec transition nodes (Methods). These include observed introductions, i.e. with clear concordance between travel-history and phylogeny (n = 205, 55.9%) and unobserved introductions (n = 162, 44.1%). Of the unobserved introductions, those that occurred before March 16th (before the drop in our sampling effort) were labeled ‘cryptic’ introductions (n = 42, 11.4%), that is, introductions where the initial travel-related case was not observed but was still likely to have occurred. Unobserved introductions after this date could not be inferred with reasonable confidence because the Québec transmission chains are currently undersampled in our sequences relative to the actual number of confirmed cases. Thus, in our analysis we only include introductions with travel-history and cryptic introductions (n = 247, Fig. 3B).
Refining the inference of introduction events using the phylogeny reduces the number of likely introduction events from 330 to 247, although both these numbers are likely to be underestimates. In particular, using information from the phylogeny reduces the number of inferred introductions from Latin America, and to a lesser extent from the USA and France, but not from the rest of Europe (Fig. 3). The inferred introduction events came from across the phylogeny of global SARS-CoV-2 diversity. Although certain parts of the phylogeny were under-represented among Québec introduction events (e.g. lineages prevalent in Asia), both phylogeny-informed and phylogeny-naive travel history captured a similar representation of the phylogeny (Fig. 3).
Fig. 3. Introductions to Québec in the global context. Circles on the phylogeny show inferred introduction events from different countries or regions (colour-coded) based on (A) travel history only (sequences with travel-history), or (B) both ancestral state reconstruction (ASR) and travel-history. Colours around the circumference of the tree show the provenance of all sequences, including those not involved in introduction events. Note that in this figure and throughout, the UK and France were considered separately from the rest of Europe.

Nine introductions were inferred to have a non-Québec to Québec transition event on the phylogeny that happened before the first reported case (Fig. 4). Of these, only one was not observed, meaning it had no known travel history. The phylogeny suggests that it was an introduction from the UK into Québec City and it places the time to the most recent common ancestor (TMRCA) of this clade as far back as January 30th. Note that this is the TMRCA of a single Québec City sequence with an English sequence, not the estimate of the date of introduction into Québec, which likely happened later (i.e. in February). The sampling date of the Québec City case was mid-March, consistent with possible cryptic transmission in Québec for an unknown period of time. Note that a study of samples from patients with flu-like symptoms between November 2019 to early March did not find any SARS-CoV-2, suggesting that introductions before late February are thought to be unlikely (as reported in Le Devoir, September 5, 2020).
Seven of the TMRCAs of the inferred introductions that fell before the first reported case on February 25th (indicated by asterisks in Fig. 4) were stem singletons in large polytomies, suggesting this part of the tree is undersampled and the TMRCA estimates should be considered with caution. These issues with large polytomies tend to happen deeper in the tree, possibly indicative of a time when fewer countries had submitted sequences. As sequencing efforts ramped up globally, the tree topology becomes better resolved.
During the spring break period, there were 26 introductions into Québec (21 observed and 5 cryptic). This is a conservative estimate since many people traveled just before and after the exact dates of spring break. Also, given the SARS-CoV-2 generation time and incubation period, some cryptic introductions during spring break are expected to appear in the counted cases past March 16th, as is observed (Fig. 4).
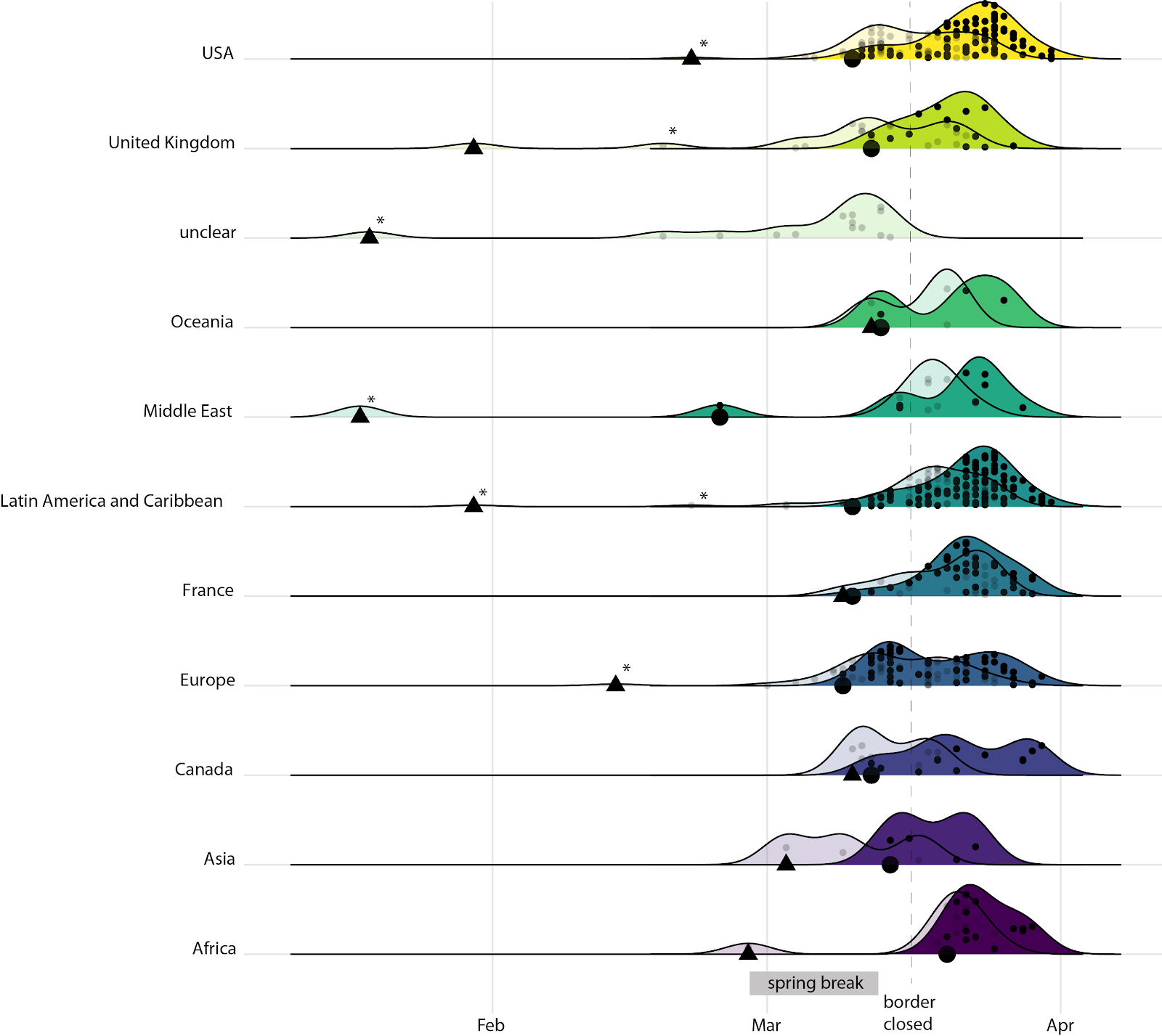
As expected, phylogeny-inferred introductions were skewed slightly earlier than the sample dates of the travel-related cases, and this skew was most pronounced for the USA, Canada, and Asia, although the latter two regions were relatively poorly represented (Fig. 4). For instance, while the bulk of the travel-related cases from the USA were sampled after the border closed, the inferred introductions suggest several cryptic introductions (11/54 introductions from the USA) happened a few days earlier, in early March (Fig. 4). Similarly, half the Canadian introductions were cryptic (4/8 introductions from other Canadian provinces). Of the cryptic introductions whose origin could not be resolved with confidence using phylogenies (labelled “unclear” in Fig. 4), all necessarily appear before the border closed because subsequent putative introductions were removed from our analysis due to undersampling of later cases in Québec. Ongoing sequencing efforts in our group aim to rectify this undersampling.
Fig. 4. Travel-related sequences and the TMRCAs of inferred introductions into Québec over time by importation region. Dark densities: small points indicate sampling dates of sequenced cases with travel history (laboratory information only). Large black points are of the first sequenced case associated with each region. Pale densities: small points indicate the TMRCA of the inferred introductions using phylogeny and travel history (thus the date of introduction into Québec will be later). Triangles are the TMRCA of the first estimated introduction from each region into Québec, based on the phylogeny. Asterisks indicate uncertainty due to stem singletons in a large polytomy.
3. SARS-CoV-2 clades introduced into Québec.
We considered two SARS-CoV-2 clade nomenclatures: Nextstrain and PANGOLIN (Rambaut et al. 2020, cov-lineages.org). Using a maximum likelihood tree of only Québec sequences, we consider which SARS-CoV-2 clades entered the various regions of Québec (Fig. 5). Among our samples, we had significantly more sequences from the Montérégie region (n = 334, compared to n = 400 from all other regions combined). This is mainly due to the fact that Montérégie was the first region to experience a spike in cases, followed by Lanaudière and Montréal. We find no examples of a particular SARS-CoV-2 clade dominating any one region in Québec, with Montérégie providing a similar sampling of clades compared to other regions (Fig. 5). This is consistent with the early cases in Québec all being travel-related, with all regions of Québec drawing on roughly the same pool of global sequences. With more cases from the growth phase of the epidemic that were contracted locally in Québec, we will have a better indication of successful establishment, spread, and any possible founder effects of SARS-CoV-2 clades in a given region.
Fig. 5. SARS-CoV-2 clades introduced into the various regions of Québec. Heatmaps correspond to two nomenclatures and are for all Québec sequences in our dataset.

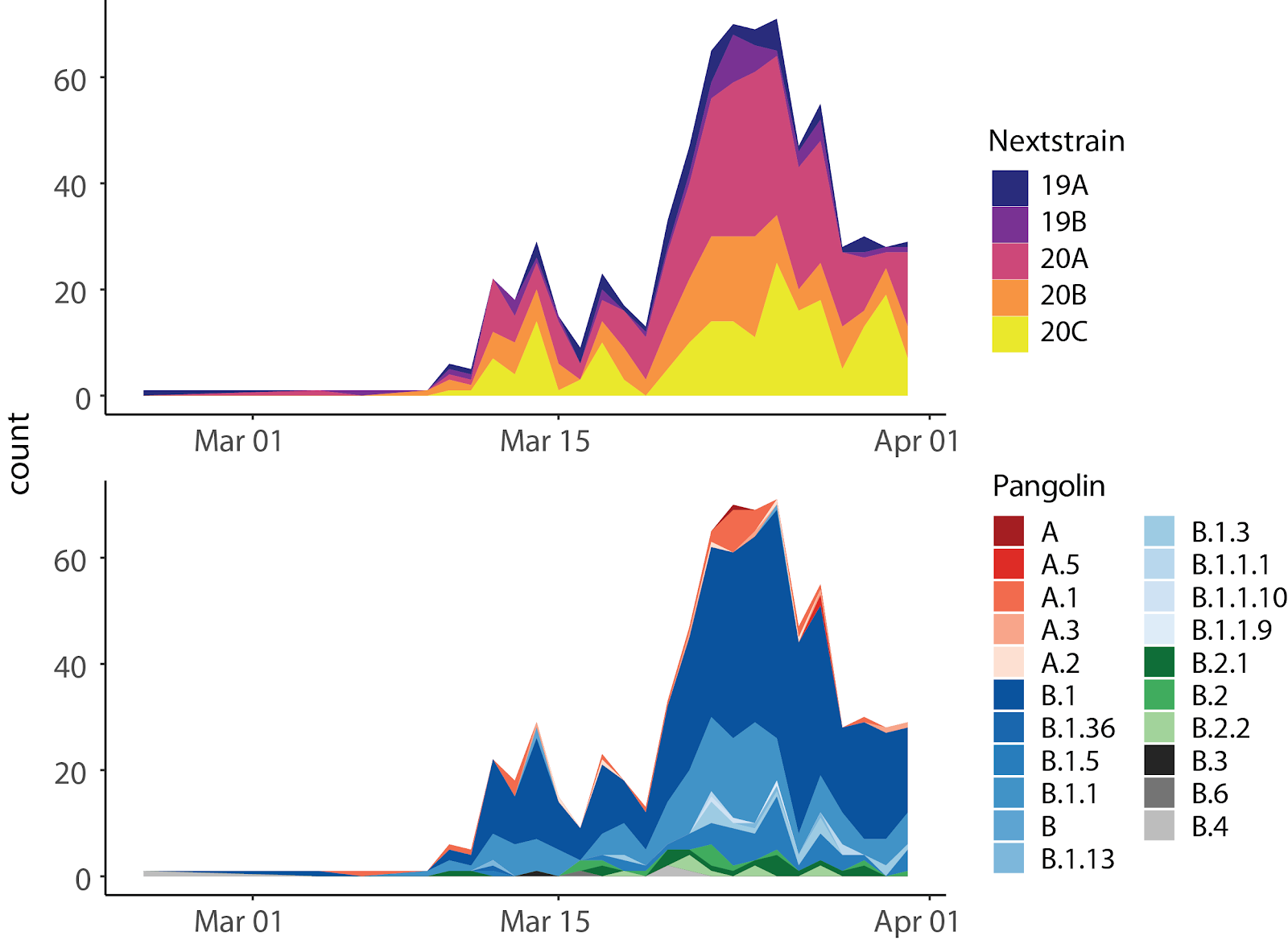
Next, we tracked the observations of different clades over time, which showed how clades B.1 and other B clades of the PANGOLIN nomenclature came to dominate the Québec epidemic by late March (Fig. 6 bottom). The early introductions of PANGOLIN clades A and B.4, common in the early outbreaks in China and Iran respectively, appear not to have been successful in Québec, as they were not observed in late March. This does not rule out possible undersampling and cryptic transmission that will be revealed in future sequencing efforts. Despite a moderate amount of travel from the UK (Fig. 3), very few of the clades common in the UK (B.3, B4, or B.6 PANGOLIN clades) were observed in Québec. Similarly, while many travel histories were linked to the USA, the major clade observed in the USA (A.3) was rare in our Québec sequences, suggesting again that in fact other lineages (mainly European ones) were being brought in via the USA during this time period. In particular, clade B.1, which originated in Italy and spread throughout Europe, became very common in Québec. The Nextstrain clade frequencies over time (Fig. 6 top) are consistent with these patterns, since the majority of sequences originate from predominantly European 2020 clades (and not 2019 clades).
Fig. 6. SARS-CoV-2 clades observed in Québec over time. Assignment of Québec sequences based on two nomenclatures: Nextstrain and PANGOLIN (cov-lineages.com). The Nextstrain clades show that the majority of Québec sequences originate from 2020 clades (20A, 20B, 20C). The PANGOLIN nomenclature shows that B.1 dominates with increasing frequency over the month of March.
Discussion
Both the epidemiological and phylogenetic analyses suggest that Québec’s early spring break, which happened before the closing of the border, played an important role in importing SARS-CoV-2 into Québec. The rest of Canada had a later spring break (or none at all due to lockdowns), and did not experience such a large spike of travel as Québec did before the border closure. Travel histories found the most common sources of COVID-19 cases to be Europe, Latin America/Caribbean, and USA, resulting in hundreds of independent introduction events into Québec, even with conservative measures. Phylogenetic analysis suggests that several of the importations from Latin America/Caribbean and the USA were more likely of European origin, and that multiple introduction events based on travel history (330) could be collapsed into fewer events (247) based on the phylogeny. Although our global context sequences were as representative as possible, it remains difficult to pinpoint precise origins of importation events due to uneven sampling across different countries. As expected, the phylogenetic analyses pushed the introduction events to slightly earlier points in time than the dates the cases were sampled (mainly after spring break). While these analyses open the possibility for several possible introduction events before the first reported case on February 25, there is significant uncertainty surrounding the inferred dates and origins of these possible introductions.
In comparison to other introduction studies, our finding that various European lineages were introduced into Québec is similar to other east coast North American outbreaks, such as New York (Gonzalez-Reiche A-S et al. 2020) and Massachusetts (Lemieux et al. 2020), and is distinct from Washington State that had introductions from China (Worobey et al. 2020). Notably, with a similar number of sampled sequences (~750), we find more importations over a shorter period of time in Québec (n > 200 from Feb 25th - April 1st) than Massachusetts (n = 80 from Jan 29th to May 9th, Lemieux et al. 2020).
This report paints a picture of the early introduction events into Québec, but our sampling falls short of comprehensively capturing the major peak in cases. Ongoing sequencing efforts will seek to determine which of the clades were most successful up to the time of the peak of cases. We have not yet included a phylodynamic analysis of these sequences, as they do not have enough temporal signal. This is likely because we have captured mostly introduction events and few within-Québec transmission chains. Additionally, the COVID-19 testing done in the early phase of the epidemic was only for those who had recently traveled or had known contact with a positive case. With more data from later March and April we hope to better capture the spread into Québec and estimate its phylodynamics.
Disclaimer: To protect patient confidentiality, in the publicly released data the first accurate date of sampling is set to March 10th. All samples taken before that are set to March 1st, and their real sampling dates are between February 25th and March 9th.
Acknowledgements
We thank all the authors, developers, and contributors to the GISAID database for making their SARS-Cov-2 sequences publicly available. We are grateful to the molecular biology team of the public health laboratory of Québec (LSPQ) including Lyne Desautels, Martine Morin and Mélanie Côté for thoroughly collecting and aliquoting all the COVID-19 positive samples. We would like to thank Marie-Michelle Simon and Patrick Willet for technical assistance on sample processing and Alexandre Belisle for automation assistance at the McGill Genome Center. Illumina and MGI sequencing was performed by Janick St-Cyr and Pierre Lepage. We thank members of the public health surveillance committee for SARS-CoV-2 for their contribution to the validation of data and their review of the manuscript and the team Immunisation et infection nosocomiale from the Public Health Institute of Quebec. The work was supported by the McGill Genome Center and the Canadian Center for Computational Genomics, two Genomics Technology Platforms (GTPs) supported by the Canadian Government through Genome Canada and a CFI grant 33408 to JR and GB. This study was also funded by a grant from Genome Canada to SM and MR under the umbrella of the Canadian COVID Genomic Network (CanCOGeN). Data analyses were enabled by compute and storage resources provided by Compute Canada and Calcul Québec.
References
Article on LSPQ study in Le Devoir: “The epidemic started around the school break” L’épidémie a commencé autour de la relâche scolaire | Le Devoir
Gonzalez-Reiche A-S, et al. (2020) Introductions and early spread of SARS-CoV-2 in the New York City area, Science, 17 Jul: Vol. 369, Issue 6501, pp. 297-301, DOI:10.1126/science.abc1917
LeBlanc JJ, Gubbay JB, Li Y, Needle R, Arneson SR, Marcino D, Charest H et al. (2020) Real-time PCR-based SARS-CoV-2 detection in Canadian laboratories. J. Clin. Virol., Jul; 128: 104433
Lemieux JE, et al. (2020) Phylogenetic analysis of SARS-CoV-2 in the Boston area highlights the role of recurrent importation and superspreading events https://www.medrxiv.org/content/10.1101/2020.08.23.20178236v1
Li C., Debruyne D.,Spencer J., et al. Highly sensitive and full-genome interrogation of SARS-CoV-2 using multiplexed PCR enrichment followed by next-generation sequencing Highly sensitive and full-genome interrogation of SARS-CoV-2 using multiplexed PCR enrichment followed by next-generation sequencing | bioRxiv
Pybus et al. (2020) Preliminary analysis of SARS-CoV-2 importation & establishment of UK transmission lineages
Québec COVID-19 data, [Accessed September 7, 2020.]
Rambaut A, Holmes EC, O’Toole Á, Hill V, McCrone JT, Ruis C, du Plessis L & Pybus OG (2020) Nature Microbiology DOI:10.1038/s41564-020-0770-5
Worobey M, et al. (2020) The emergence of SARS-CoV-2 in Europe and North America, Science, 10 Sep:eabc8169, DOI: 10.1126/science.abc8169