Increasing frequency of the P.1 lineage in Manaus
Following up on our previous post, here we share a frequency table by date of collection for a total of 142 SARS-CoV-2 genome sequences from Manaus, including 115 partial, near-complete, and complete SARS-CoV-2 genome sequences generated by our team from samples collected in December 2020 (n=67, collection dates between 15 December 2020 and 31 December 2020) and January 2021 (n=48, collection dates between 1 Jan 2021 and 9 Jan 2021). Near-complete and complete sequences were classified using pangolin; partial sequences were typed using maximum likelihood phylogenetic analysis with an in-house dataset of near-complete or complete P.1, P.2, and B.1.1.28 sequences.
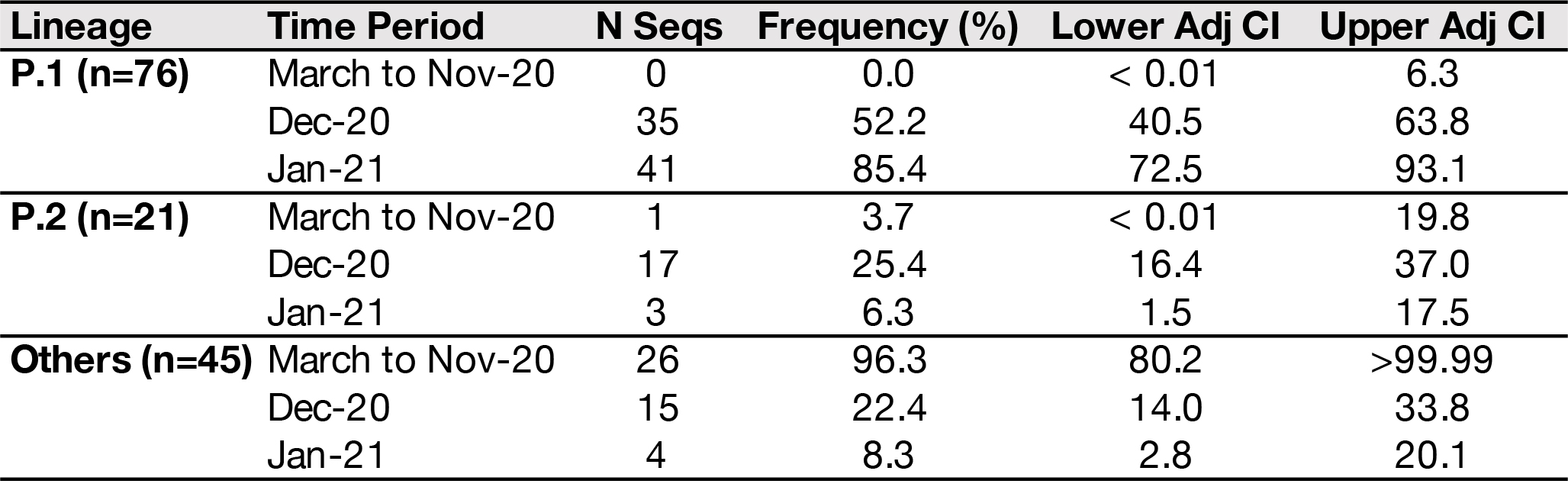
Our data suggest a recent increase in the proportion of P.1 cases in Manaus (Table 1). The P.1 lineage was not detected in Manaus between March and November. However, we found that 52.2% (n=35/67) of the SARS-CoV-2 typed cases from December were caused by the P.1 lineage. In January 2021, we detected an increase of P.1 lineage frequency to 85.4 % (n=41/48). We also detected a modest increase in the frequency of P.2 cases in December 2020 to 25.4% (n=17/67), but fewer P.2 cases were detected in January 2021. The frequency of other lineages decreased from 96.3% between March and November 2020 to 8.3% in January 2021.
Table 1. Frequency table of lineage distribution in Manaus by period of sample collection (n=142). N Seqs = number of genotyped samples. CI = confidence interval estimated using the adjusted Wald method.
This analysis includes considerably more data compared to our previous post on the recent emergence of a new P.1 lineage in Manaus and suggests that most recent cases in Manaus are being caused by local transmission of the P.1 lineage, although P.2 and other lineages might still be circulating. These results should be considered preliminary at this stage. Larger representative datasets are required to investigate in more detail changes in the frequency of this lineage in Manaus and elsewhere.
For more information and details please contact Dr. Nuno R. Faria ([email protected]) and Dr. Ester Sabino ([email protected]).