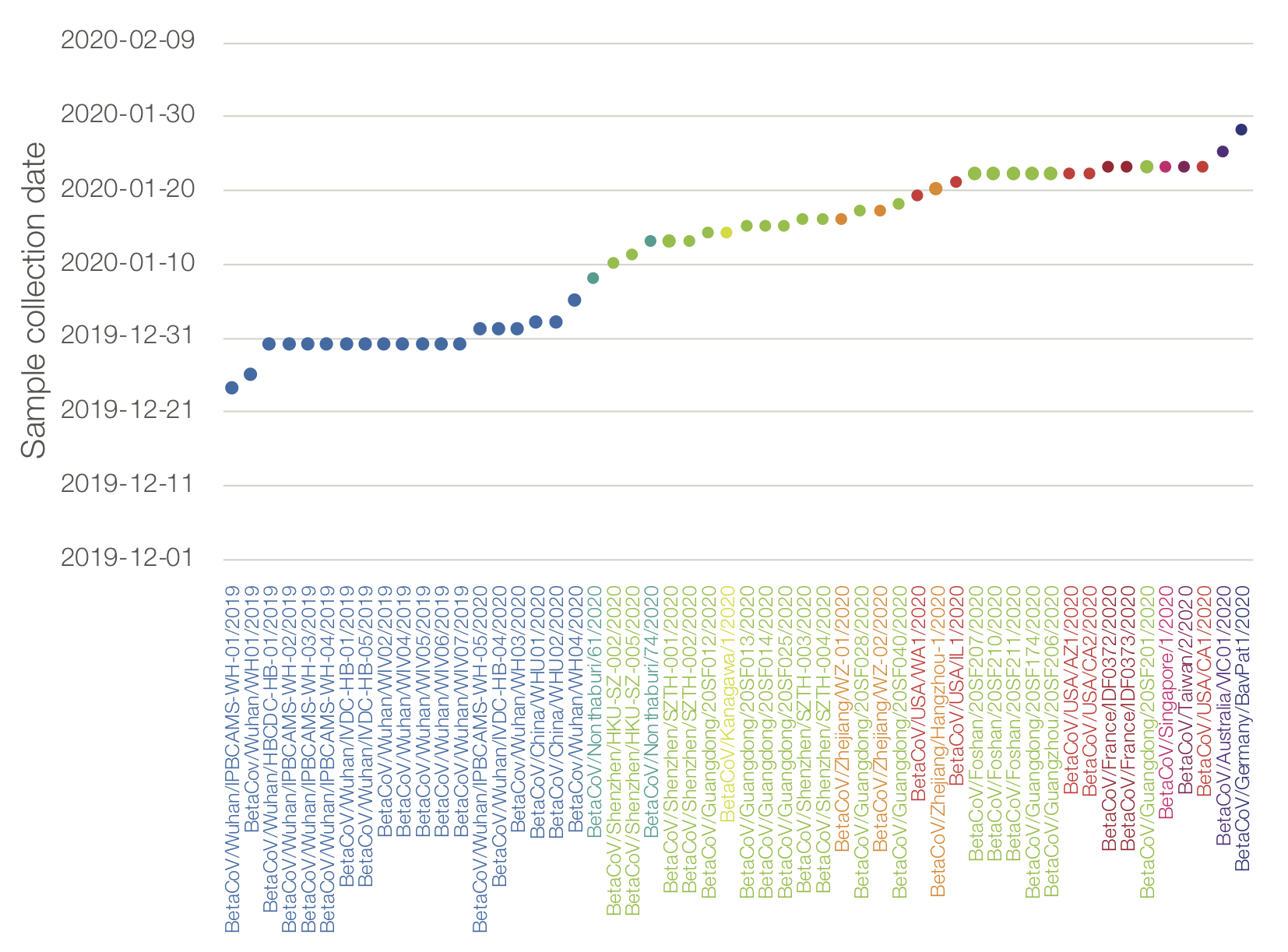
Genome sequences as of 02/02/2020. Dots represent date of collection of the sample. Colour by location of case.
1 Like
I’m really concerned about the lack of genome sequences from Hubei after Jan 5 (or any new sequences from China in the last week). The long branch to the second California genome shows that there is likely a lot of unsampled diversity out there.
What would be the best way to go about accounting for population structure? I don’t think it’s practical to model each province of China and each country as a separate deme. Could we consider sequences from patients not sampled in Hubei, but likely infected in Hubei, as part of a well-mixed Hubei epidemic? How hard would it be to add a restriction that prevents them from coalescing with any other sequence after the patient left Hubei (assuming we have this metadata).
I think we just have to wait for more data. But yes - I am going through all the reports to look for travel history. Most have Wuhan in their travel history but I suspect that is how they are being picked up.