First reported cases of SARS-CoV-2 sub-lineage B.1.617.2 in Brazil: an outbreak in
a ship and alert for spread
Mirleide Cordeiro dos Santos 1, Edivaldo Costa Sousa Júnior 1, Jessylene de Almeida
Ferreira 1, Luana Soares Barbagelata 1, Sandro Patroca da Silva 2, Amanda Mendes Silva 1,
Jedson Cardoso 1, Rayssa Layna da Silva Bedran 1, Wanderley Dias das Chagas Junior 1,
Delana Andreza Melo Bezerra 1, Kenny da Costa Pinheiro 1, Dielle Teixeira Monteiro 1,
Patrícia dos Santos Lobo 1, Gabriela Medeiros Muniz 1, James Lima Ferreira 1, Luis Felipe
Lima Lobato3, Carlos Eduardo Campos Maramaldo 3, Hivylla Lorrane dos Santos Ferreira 3, Lídio Gonçalves Lima Neto 3, Antônio Carlos Rosário Vallinoto 4, Ana Cecília Ribeiro Cruz 2, Daniele Barbosa de Almeida Medeiros 2, Luana Silva Soares 1*
1 Department of Virology, Evandro Chagas Institute, Highway BR-316, km-07, s/n,
Ananindeua, Para state, Brazil zipcode 67030-000.
2 Department of Arbovirology and Hemorrhagic Fevers, Evandro Chagas Institute,
Highway BR-316, km-07, s/n, Ananindeua, Para state, Brazil zipcode 67030-000.
3 Central Laboratory of Public Health of the State Maranhão.
4 Virology Laboratory, Federal University of Pará.
*Corresponding authors:
Mirleide Cordeiro dos Santos
Phone:+55(91) 3214-2007 E-mail: [email protected]
Luana Silva Soares
Phone:+55(91) 3214-2007 E-mail: [email protected].
SUMMARY
The variant B.1.617 emerged in India in late 2020 and diversified into three genetically distinct sub-lineages (B.1.617.1, B.1.617.2 and B.1.617. 3). However, sub-lineage B.1.617.2 has stood out due to the sharp increase in the number of cases associated with an increasing proportion of sequenced viruses belonging to this sub-lineage. Therefore, it is considered a variant of concern. In this context, targeted genomic surveillance must be improved (including cases associated with travel, clusters or outbreaks) in order to understand and inform the potential implications of this variant for public health. Thus, the objective was to report the preliminary results of the first identification of variant B.1.617.2 of SARS-CoV-2 isolated from cases imported into Brazil. For this purpose, 24 crew members of a ship of Asian origin that docked in Brazil in May 2021 were investigated, of these 15 were positive in RT-qPCR and six were subjected to genomic sequencing by the MiniSeq - Illumina Platform, then the analysis of bioinformatics and the submission of the generated sequences to the Pangolin platform (Phylogenetic
Assignment Of Named Global Outbreak Lineages) v 2.4.2, for the classification of the strains detected in the sequenced samples. The results obtained allowed to identify the occurrence of the sub-lineage B.1.617.2 of SARS-CoV-2, where all sequences share 21 amino acid changes in the Spike protein. In this sense, further characterization of these strains is necessary to allow a complete assessment of their potential implications for public health.
Keywords: Phylogenetic Variants, Outbreak, COVID-19
INTRODUCTION
COVID-19 remains a constant concern for the population and the competent authorities despite intense surveillance, advances in vaccinology, pharmaceutical and non-pharmaceutical interventions. Although all the aforementioned scientific efforts, its rapid dissemination has contributed to the diversification in several strains phylogenetics marked by different point mutations that reflect current transmission currents (Fontanet et al., 2021). In this scenery, a sensitive view to be considered is the export/import of new variants of SARS-CoV-2 by ship trades, once the risk of transmission increases due to the number of days that the crew members spend on board, favoring the occurrence of outbreaks (Guagliardo et al., 2021). These events require rapid notification to prevent the spread of strains from other locations.
The B.1.617 strain of SARS-CoV-2 was first reported in October 2020 in India and it has been associated with an increase in the number of daily infections in this country (Ferreira et al., 2021). Genetically, the B.1.617 strain has particular signature in the protein Spike, characterized by six mutations: D111D, G142D, L452R, E484Q, D614G, and P681R (Cherian et al., 2021). Initially, it was designated as a Variant of Interest (VOI) by WHO (WHO, 2021). However, on May 10 of 2021, it was reclassified as a Variant of Concern (VOC) considering the recent information on its transmissibility and potential for neutralizing antibodies (Hoffmann et al., 2021, Yadav et al., 2021a, Yadav, et al., 2021b).
The B.1.617 strain of SARS-CoV-2 has been reported in more than 51 countries and now in all continents. Until this report, this lineage shows three distinct sub-lineages: B.1.617.1, B.1.617.2 and B.1.617.3, which the difference among them is in Spike protein (Cherian et al., 2021, Hoffmann et al., 2021). Global genomic surveillance conducted by WHO using the sequences available in GISAID (Global Initiative on Sharing All Influenza Database), indicates a substantial increase in the circulation of sub-lineage B.1.617.2 (WHO, 2021). There are many factors still unclear about variant B.1.617.2, so
the rapid response of surveillance combining genetic and epidemiological data to investigate the genetic diversity, evolution and epidemiology of SARS-CoV-2 variants is of utmost importance considering its impact in public health. Here, we report our preliminary results from the investigation of COVID-19 of Asiatic origin ship that docked in Brazil, related to SARS-CoV-2 variants B.1.617.2 This finding brings the alert to SARS-CoV-2 variants B.1.617.2 spread in South America and its impact on world public health.
METHODS
On May 13 of 2021, National Health Surveillance Agency (ANVISA) notified a COVID-19 suspect case among the crew members of the ship “MV SHANDONG DA ZHI” that docked at the port in Maranhão State. Central Public Health Laboratory (LACEN-MA) of the Secretary of Health Surveillance of Maranhão State (SHS-MA) lead the sample collection through nasopharyngeal swab from 24 crew member.
The laboratory diagnosis was performed simultaneously by LACEN-MA and Laboratory of Respiratory Viruses of the Evandro Chagas Institute (LVR/IEC; The Brazilian Regional Reference Laboratory located in Ananindeua city, Para State), using RT-qPCR assay following the guidelines of the protocol provided by the Atlanta CDC (CDC, 2020). The positive samples with Ct < 25 were conducted to viral lineage investigation by Next Generation Sequencing. The library preparation was performed using the amplicon methodology using the CleanPlex® SARS-CoV-2 Research and Surveillance Panels kit
(ref. 918010), and sequenced in the MiniSeq, Illumina Platform. Preprocess began removing sequences of adapters, reads with low quality and less than 36 bp size, using Trimmommatic program (7). For Trimmomatic, we have used the following parameters: LEADING: 3 TRAILING: 3 MINLEN: 26.
The reference mapping and prediction of open reading frames (ORFs) and functional annotation were automatically performed with Geneious v.8.1.9 (8) (identity, 99.96%) using a reference sequence from NCBI database (NC_045512). The genome was manually curated by comparison of the coding ORFs with this genome using Geneious v.8.1.9 (8). The genomes were classified by software Pangolin v.2.4.2 (GitHub - cov-lineages/pangolin: Software package for assigning SARS-CoV-2 genome sequences to global lineages.) (9). All tools were used with default parameters unless otherwise specified. Additional analyses were performed using SOPHiA GENETICS software (https://www.sophiagenetics.com) to assemble and identification of new variants.
RESULTS
On March 27 of 2021, The internationally-operated “MV SHANDONG DA ZHI” ship left Malaysia, initiating its journey to Wood of Port, in São Luis city, Maranhão State, Brazil. On its way, this ship docked in Cape Town, South Africa, when 24 crew members on boarded on April 21. They arrived in Brazil on May 14, notifying suspicion of one COVID-19 case to the National Health Surveillance Agency (ANVISA) and Brazilian Ministry of Health as well (Figure 1).
Figure 1. Path taken by the ship MV SHANDONG DA ZHI.
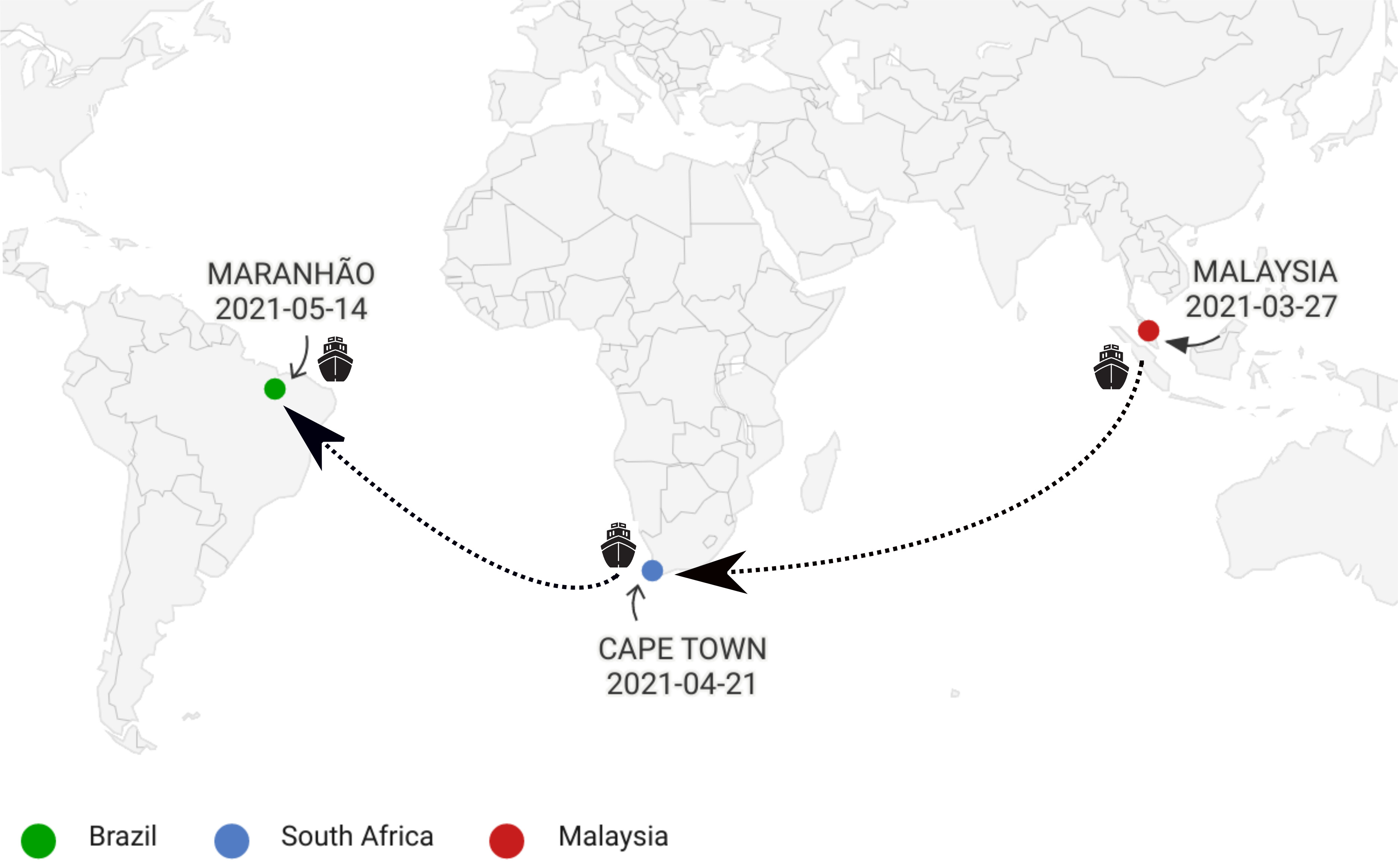
The index case was a 54-year-old Indian man showed respiratory symptoms compatible to COVID-19, starting on May 4. On May 13, the diagnosis was confirmed as a COVID-19 case by LACEN-MA and LVR/IEC, base in RT-qPCR. For this reason, an investigation was started, including all contacts in the ship and the respiratory secretion was collected by LACEN-MA on May 16. The samples were sent for genomic sequencing at Evandro Chagas Institute.
From 24 crew members, 15 (15/24; 62.5%) were positive to SARS-CoV-2 detection. From these, three (3/15; 60%) were symptomatic. Just the index case was hospitalized in an Intensive Care Unit (ICU) of a private hospital in São Luis city, capital of Maranhão State. All crew members have been isolated, under daily medical monitoring. Six sample reached the Ct criteria to NGS requirement. SARS-CoV-2 genomes were obtained and classified as a lineage B.1.617, conformed by Pangolin lineage classification. The whole-genome sequence of the SARS-CoV-2 was obtained and the average coverage depth 18,300x reads (extending from 7,540x to 25,700x).
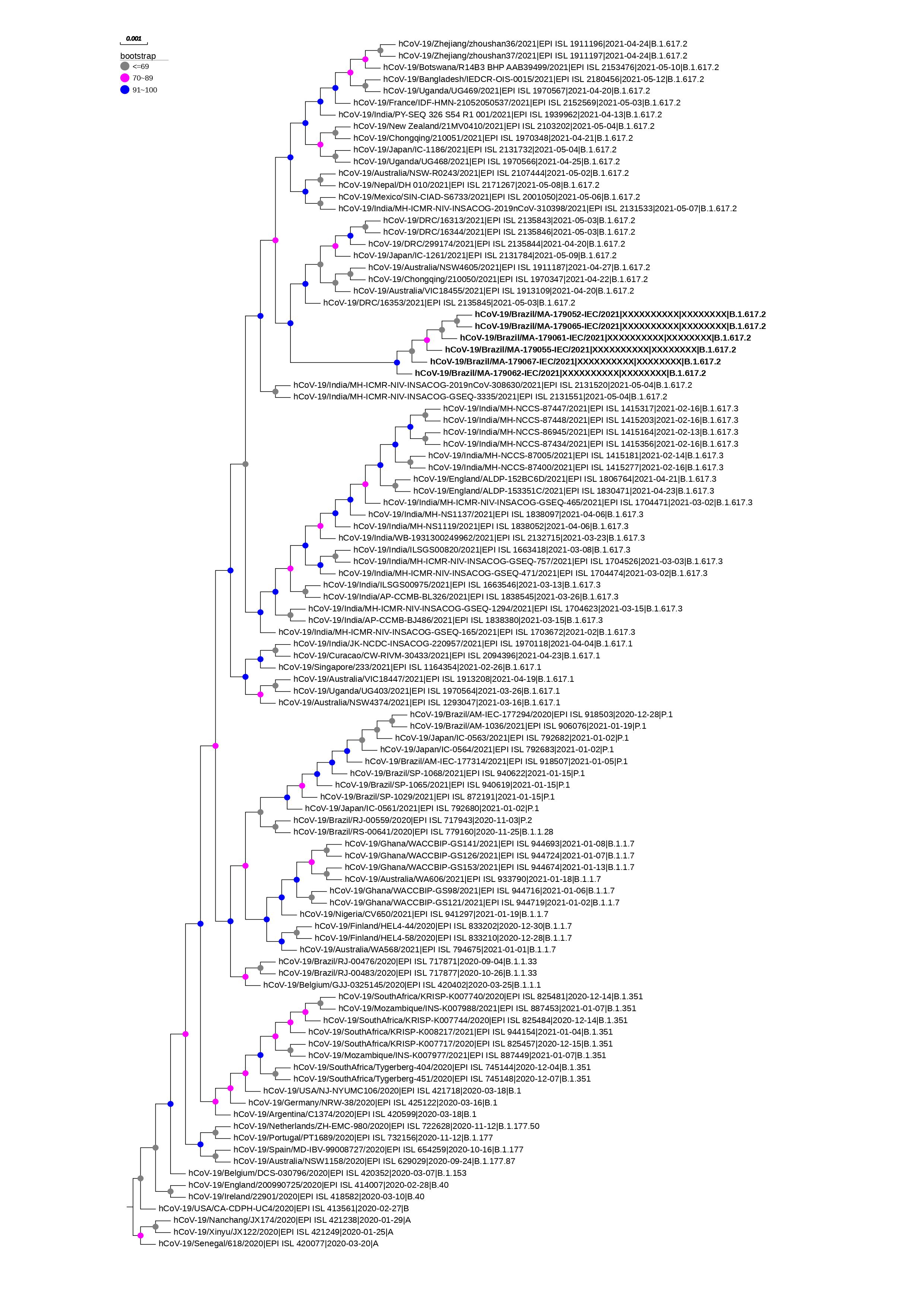
Concerning aminoacid mutation on Spike protein, the changes was in T19R, L452R, T478K, P681R, and D950N positions, which consist to genetic signature of this sub-lineage. In addition to the marker mutations of the B.617.2 strain, 21 changes in the Spike protein were observed being shared between the sequences (Table 1). Of which, phylogenetic data showed that these sequences are grouped as a monophyletic group (figure 2).
Table 1. Amino acidic changes located in the Spike protein of a B.1.617.2 sequence. The yellow markings correspond to the genetic markers of sub-lineage B.1.617.2.

Figure 2. Phylogenetic tree of SARS-CoV-2 strains isolated from an outbreak caused by lineage B.1.617.2 on a ship anchored in the State of Maranhão, Brazil. The gray, pink and blue circles indicate the bootstrap values.
CONCLUSION
Here we report the early detection of the SARS-CoV-2 lineage B.1.617 in Brazil associated to imported cases. This single outbreak reinforces the necessity of continuous monitoring of travelers to follow the real-time spread of this new SARS-CoV-2 variants, which has genetic marks probably related to evolutionary selective advantages that could impact public health as well as the immunization strategies.
ACKNOWLEDGEMENTS
The authors would like to thank all the professionals who worked bravely to deal with this pandemic. We thank the Evandro Chagas Institute, where the development of the research was carried out with great contribution from the virology team. We would also like to thank the General Coordination of Laboratories (CGLab) of the Ministry of Health (MS), Central Public Health Laboratory (LACEN-MA) and Secretary of Health Surveillance of Maranhão State (SHS-MA).
FINAL SUPPORT
This study was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
REFERENCES
Bolger, M. et al. “Trimmomatic: a flexible trimmer for Illumina sequence data.” Bioinformatics 30.15 (2014): 2114-2120.
Center dor Disease Control and Prevention. CDC 2019-Novel Coronavirus (2019-nCoV) Real-Time RT-PCR Diagnostic Panel . [Online] US Centers for Disease Control and Prevention. [Accessed 25 May 2021]. (2020)
Cherian, S. et al. Convergent evolution of SARS-CoV-2 spike mutations, L452R, E484Q and P681R, in the second wave of COVID-19 in Maharashtra, India. Preprint at bioRxiv Convergent evolution of SARS-CoV-2 spike mutations, L452R, E484Q and P681R, in the second wave of COVID-19 in Maharashtra, India | bioRxiv (2021).
Ferreira, I. et al. SARS-CoV-2 B.1.617 emergence and sensitivity to vaccine elicited antibodies. Preprint at bioRxiv https://doi.org/10.1101/2021.05.08.443253 (2021).
Hoffmann, M. et al. SARS-CoV-2 variant B.1.617 is resistant to Bamlanivimab and evades antibodies induced by infection and vaccinatio. Preprint at bioRxiv https://doi.org/10.1101/2021.05.04.442663 (2021).
Kearse, M. et al. “Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data.” Bioinformatics 28.12 (2012): 1647-1649.
WHO - COVID-19 Weekly Epidemiological Update, 9 may 2021 at https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19--
-11-may-2021
Yadav, P. D. et al. SARS CoV-2 variant B.1.617.1 is highly pathogenic in hamsters than
B.1 variant. Preprint at bioRxiv https://doi.org/10.1101/2021.05.05.442760 (2021a).
Yadav, P. D. et al. Neutralization of variant under investigation B.1.617 with sera of
BBV152 vaccinees. Clin. Infect. Dis. Neutralization of Variant Under Investigation B.1.617.1 With Sera of BBV152 Vaccinees | Clinical Infectious Diseases | Oxford Academic (2021b).