Markus H. Antwerpen, Daniel Lang, Sabine Zange, Mathias C. Walter* and Roman Wölfel
Bundeswehr Institute of Microbiology, Munich, Germany
*contact: [email protected]
Since early May 2022, dozens of suspected and confirmed monkeypox infections have been reported in several European and North American countries. The first German monkeypox case was reported in Munich on May 19, where the 26 year-old patient had shown characteristic skin changes. During his travel through Europe, he finally showed mild symptoms and requested medical examination.
The Bundeswehr Institute of Microbiology performed primary diagnostics of a swab taken from a skin lesion and subsequently sequenced the sample. Here, we announce the available data to the scientific community.
DNA extraction was performed using DNeasy Mini Kit (Qiagen, Hilden, Germany) from clinical material and eluted in 100µl EB-Buffer. DNA concentrations were quantified using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Dreieich, Germany) according to the manufacturers’ protocols. From total DNA, an Illumina-compatible library was prepared (NEBNext® Ultra™ II DNA Library Prep Kit, NEB, Frankfurt am Main, Germany) and sequenced on a MiSeq instrument (Illumina, San Diego, CA, USA) using 2x 150bp v2 chemistry in order to obtained paired-end reads.
Raw reads were assigned by Kraken 2 [1] and human reads were discarded. Remaining paired-end reads were assembled de-novo using an in-house script based on the SPAdes assembler [2] in single-cell mode. In addition, viral reads were mapped to MPXV_USA_2022_MA001 (AccNo:ON563414) for validation and manual curation of the reported genome sequence. Afterwards, a contig extension was performed using the previously assembled contigs.
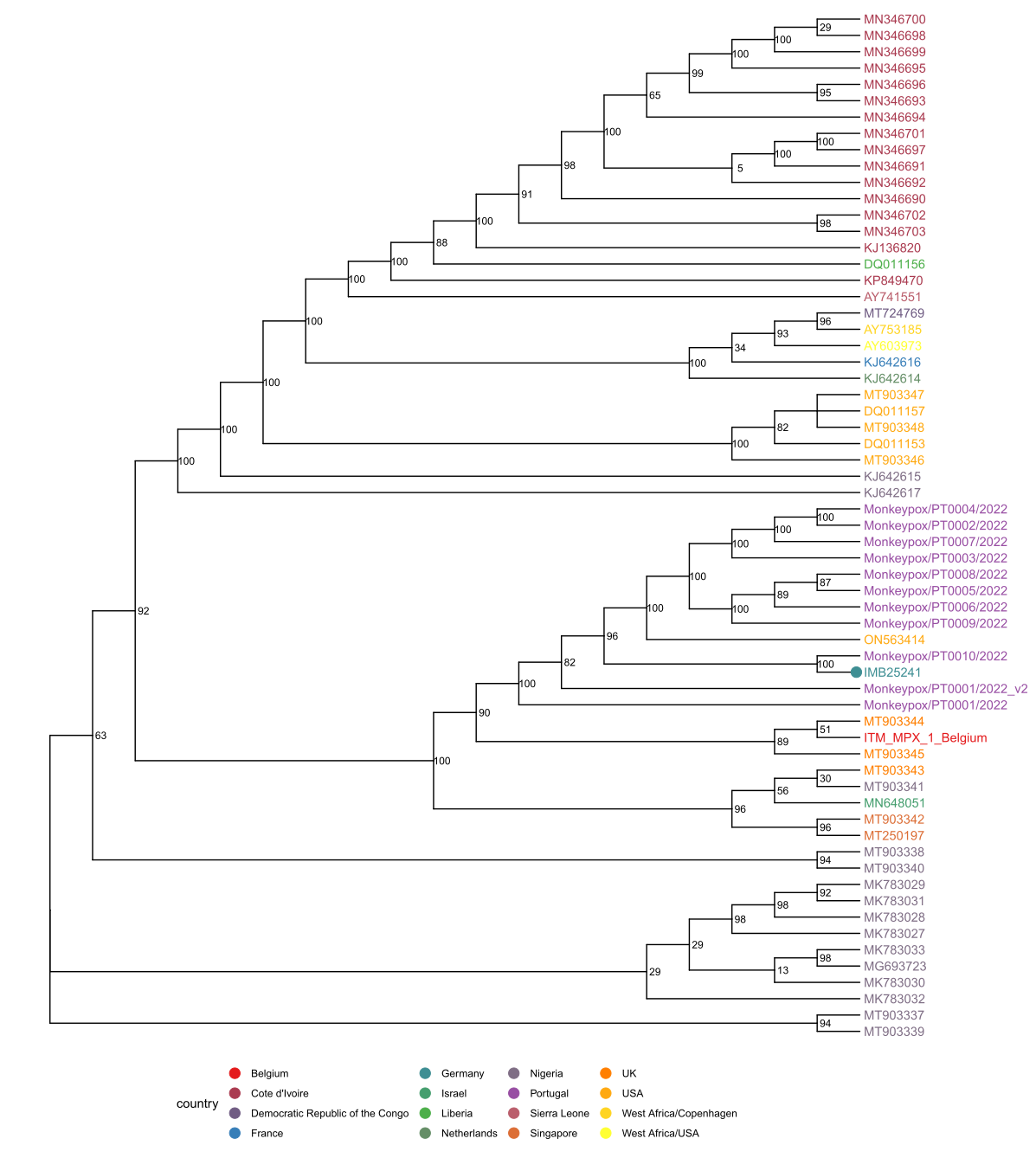
The full-length genome comprises 197.378 bp and was sequenced directly from clinical material. BLAST analysis [3] and phylogenetic inference support the classification of this isolate into the West-African clade associated with the recent isolates from Europe and the US. The nearest neighbor is PT0010/2022, an isolate from Portugal (published May 23 by INSA). All of the identified SNPs compared to the MPXV_UK_P2 are either TC→TT or GA→AA in dinucleotide context and potentially caused by APOBEC3 as hypothesized by Rambaut. In addition, a 10 bp deletion (CAATCTTTCT) was discovered at 133.175 bp which is part of an exact tandem repeat or an inexact triplet repeat upstream of a hypothetical protein. Interestingly, this duplication is not annotated in the recently published CDC strain ON563414 and the Belgium strain ITM_MPX_1_Belgium but in all sequences from Portugal (PT0001-PT0009).
The genome sequence was submitted to NCBI Nucleotide (ON568298) and will be updated (if needed) as soon as data from the grown cell-cultures are available.
Phylogenetic analysis is based on an incremental analysis of all publicly available full-length (>180 kbp) sequences acquired via NCBI Nucleotide or shared here by colleagues on virological.org. Recombinant or apparently partially older sequences were discarded. Multiple sequence alignments were carried out using MAFFT v7.490 (options: --auto --6merpair; [4]). Phylogenetic inference with maximum likelihood as implemented in fasttree [5] using the GTRCAT model with subsequent rescaling of branch lengths optimizing a discrete gamma model with 20 rate categories (fasttreemp option: -nt -gtr --gamma; binary compiled with -DUSE_DOUBLE). The full tree was rooted at the separation between the Central and West African clades and sequences of the West African Clade were selected for a reanalysis that included also the most recent sequences from Portugal. It was rooted at the midpoint and is shown below.
References
- D. E. Wood, J. Lu, and B. Langmead, “Improved metagenomic analysis with Kraken 2” Genome Biol., vol. 20, no. 1, p. 257, Nov. 2019, doi: 10.1186/s13059-019-1891-0.
- A. Bankevich et al., “SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing.” J Comput Biol, vol. 19, no. 5, pp. 455–477, May 2012, doi: 10.1089/cmb.2012.0021.
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers, and D. J. Lipman, “Basic local alignment search tool.” J Mol Biol, vol. 215, no. 3, pp. 403–410, Oct. 1990, doi: 10.1006/jmbi.1990.9999.
- K. Katoh, K. Misawa, K. Kuma, and T. Miyata, “MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform.” Nucleic Acids Res, vol. 30, no. 14, pp. 3059–3066, Jul. 2002.
- M. N. Price, P. S. Dehal, and A. P. Arkin, “FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix” Mol. Biol. Evol., vol. 26, no. 7, pp. 1641–1650, Jul. 2009, doi: 10.1093/molbev/msp077.