Exploring the natural origins of SARS-CoV-2
Spyros Lytras1, Joseph Hughes1, Xiaowei Jiang2, David L Robertson1
1MRC-University of Glasgow Centre for Virus Research (CVR), Glasgow, UK.
2Department of Biological Sciences, Xi’an Jiaotong-Liverpool University (XJTLU), Suzhou, China.
A longer version of this text is now available as a preprint: Exploring the natural origins of SARS-CoV-2 in the light of recombination | bioRxiv
The lack of an identifiable intermediate host species for the proximal animal ancestor of SARS-CoV-2 and the distance (~1500 km) from Wuhan to Yunnan province, where the closest evolutionary related coronaviruses circulating in horseshoe bats have been identified, is fueling speculation on the natural origins of SARS-CoV-2. Here we put this distance into the context of the geographical ranges of potential bat hosts across China.
SARS-CoV-2 is a member of the Sarbecovirus subgenus of Betacoronaviruses found in horseshoe bat hosts (family Rhinophilidae) and a sister lineage of SARS-CoV (Figure 1A), the causative agent of the SARS outbreak in 2002-3 (Gorbalenya et al. 2020). We have performed recombination detection analysis on a whole genome alignment of all the available Sarbecoviruses , focusing on the broader set of ‘nCoV’ viruses that cluster with SARS-CoV-2 in phylogenetic analysis (Figure 1A). This identified 16 recombination breakpoints that can be used to split the alignment into 17 putatively non-recombinant genomic regions from which a phylogeny can be inferred. To clearly characterise the recombination patterns between viruses in the same clade as SARS-CoV-2 and those in the sister lineage, which includes SARS-CoV, we have attributed each virus in each of the 17 regions to either being in the nCoV clade (closer to SARS-CoV-2) or the non-nCoV clade (closer to SARS-CoV) (Figure 1A), similarly defined in MacLean et al. (2020).
While the two genetically closest relatives to SARS-CoV-2 identified so far are the bat Sarbecoviruses RaTG13 and RmYN02 (Zhou et al. 2020; Zhou et al. 2020), both recombinants from samples collected in Yunnan (Figure 1B), they are estimated to have shared a common ancestor with SARS-CoV-2 about 40/50 years ago (Boni et al. 2020; Wang, Pipes, and Nielsen 2020; MacLean et al. 2020) so are too distant to be SARS-CoV-2’s progenitors. Importantly, three recombinant bat Sarbecoviruses, CoVZC45, CoVZXC21 and Longquan_140, the next closest SARS-CoV-2 relatives in the nCoV clade (for most of their genomes for CoVZC45 and CoVZXC21, except for four regions on Orf1ab and Spike, and for two parts of Longquan_140’s genome) were all found in Zhejiang a coastal province in Eastern China (Hu et al. 2018; Lin et al. 2017) (Figure 1B).
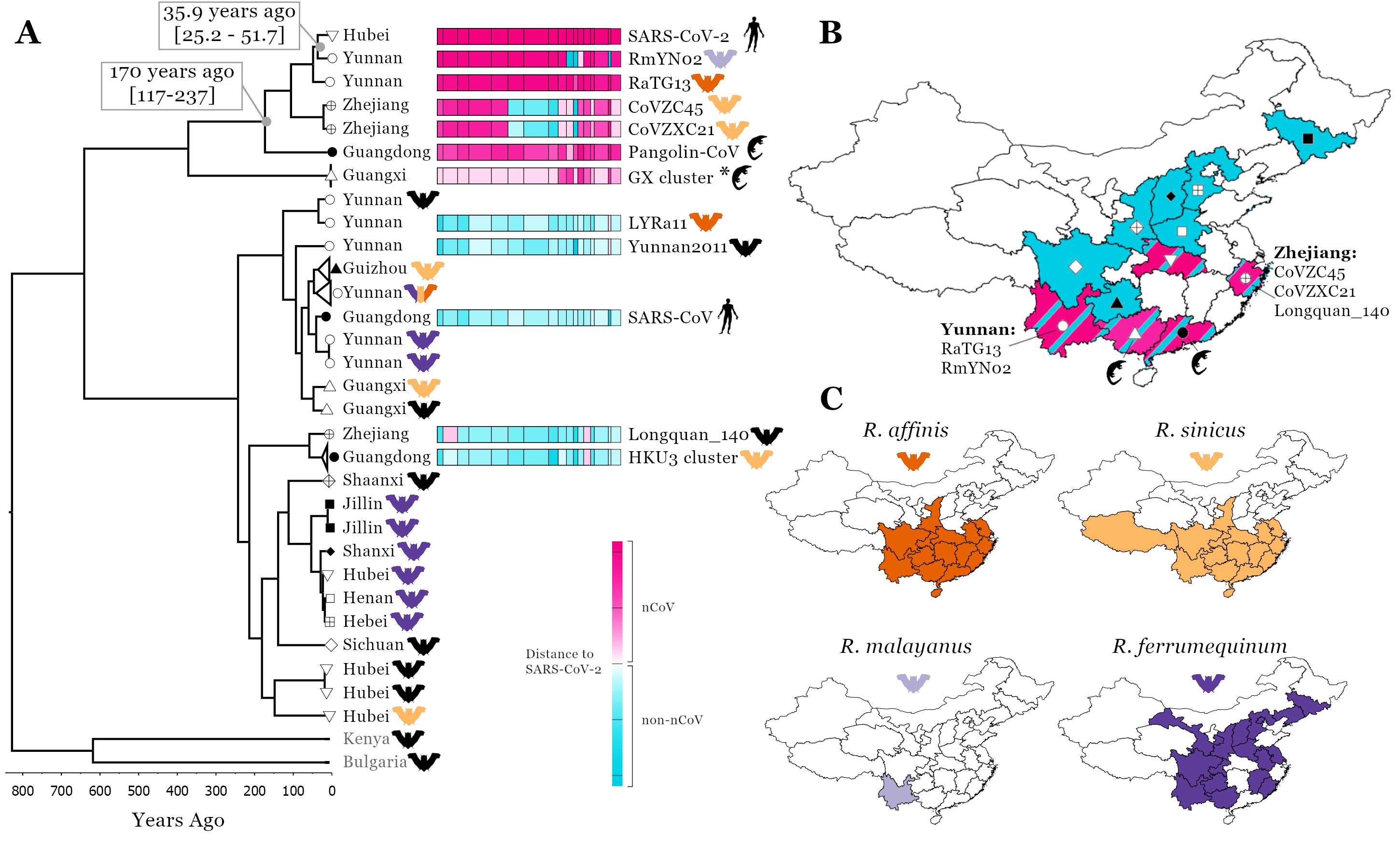
Figure 1. Linking SARS-CoV-2 related Sarbecovirus to geography and host ranges. Recombination analysis on a whole genome alignment of 69 Sarbecoviruses identified 16 recombination breakpoints [A], see methods. The fourth region was used for a molecular clock analysis (see scale below phylogenetic tree, a representative set of 32 viruses are shown) and the median node age is presented for two key nodes including 95% HPD intervals.To illustrate recombination patterns between viruses in the same clade as SARS-CoV-2 and those in other clades of the tree we have attributed each region of each virus to either being in the nCoV clade (the clade SARS-CoV-2 is found in, pink) or the non-nCoV clade (closer to SARS-CoV, blue). The colour shade corresponds to genetic distance, see key. A map of China is shown with colours corresponding to the regions Sarbecoviruses have been collected [B]: blue, the non-nCoV clade and pink, the nCoV clade. Symbols correspond to provinces at tree tips in A. Host ranges of four potential hosts of the proximal SARS-CoV-2 ancestor [C]. Ranges from Smith and Xie (2013). Geographical and genomic visualisation was performed using D3 and JavaScript in Observable.
This high prevalence of recombination, the bringing together of evolutionary divergent genome regions in co-infected hosts to form a hybrid virus, is typical of many RNA viruses and for coronaviruses provides a balance to their relatively slow evolutionary rate (Graham and Baric 2010). Recombinants with parts of their genomes shared with the SARS-CoV-2 progenitor (between 40 and about 100 years ago, Figure 1A) are distributed on both sides of China (a distance of ~2000 km) indicating the urgent need to broaden the geographical region being searched for the SARS-CoV-2’s immediate animal ancestor and avoiding being overly focussed on the Yunnan location of the two closest Sarbecoviruses RaTG13 and RmYN02.
Furthermore, the finding that Malayan pangolins, Manis javanica, non-native to China, the other mammal species from which Sarbecoviruses related to SARS-CoV-2 have been sampled from in Guangxi and Guangdong provinces in the southern part of China (Lam et al. 2020; Xiao et al. 2020), indicates these animals are being infected in this part of the country. Pangolins are one of the most frequently trafficked animals with multiple smuggling routes leading to Southern China (Xu et al. 2016). The most common routes involve moving the animals from Southeast Asia (Myanmar, Malaysia, Laos, Indonesia, Vietnam) to Guangxi, Guangdong, and Yunnan. The most likely scenario is that Sarbecoviruses infected the pangolins after they were trafficked into Southern China, consistent with the respiratory distress they exhibit (Liu, Chen, and Chen 2019; Xiao et al. 2020). Although, the recent confirmation of a Sarbecovirus sampled from a Chinese pangolin, Manis pentadactyla, in Yunnan, collected in 2017 (GISAID ID EPI_ISL_610156, authors: Jian-Bo Li, Hang Liu, Ting-Ting Yin, Min-Sheng Peng and Ya-Ping Zhang of the State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences), does raise the question, are Chinese pangolins infected endemically? The key, and urgent question to minimise future spillovers, is thus not, how did SARS-CoV-2 get from Yunnan to Hubei, but rather which bat or other Chinese animal species are harbouring nCoV Sarbecoviruses?
Of the horseshoe bat species that have their ranges sufficiently dispersed across China to account for this geographical spread: bat Sarbecovirus recombinants in the West and East China, imported pangolins and bat Sarbecovirus recombinant links to Southern China and SARS-CoV-2 emergence towards the north in Hubei (Figure 1B); suggests particular focus should be placed on R. affinis and R.sinicus. Strikingly, the ranges of these species are almost perfectly overlapping, especially for R. affinis and R. sinicus across the regions of China where all the nCoV viruses have been collected (Figure 1C). R. ferrumequinum is not found in large parts of Central or Southern China, while R. malayanus is found in the West part of China only.
Conclusion. The currently available data, although sparse, illustrates a complex history behind the natural evolution of SARS-CoV-2, governed by co-circulation of related coronaviruses, over at least the last 100 years, across the bat populations from East-to-West/Central and Southern China and multiple recombination events imprinted on the genomes of these viruses. Having presented evidence in support for R. affinis’s importance, it should be noted at least 20 different Rhinolophus species are distributed across China (four being endemic to China) leaving many species for which the viruses are unknown. The risk of future emergence of a new SARS-CoV-2 nCoV strain is too high to restrict sampling strategies.
Acknowledgements
We thank all the researchers who have kindly deposited and shared genome data on GISAID. Credit also needs to be given to the surveillance projects for generating the genome data that is available in GenBank and to the software developers for making the tools we have used freely available. DLR and JH are funded by the MRC (MC_UU_1201412). SL is funded by an MRC studentship.
Methods
The whole genome sequences of 69 Sarbecoviruses were aligned and the open reading frames (ORF) of the major protein-coding genes were defined based on SARS-CoV-2 annotation. To minimise alignment error codon-level alignments of the ORFs were created using MAFFT (Katoh et al. 2005) and Pal2Nal (Suyama, Torrents, and Bork 2006). The intergenic regions were also aligned separately using MAFFT and all alignments were pieced together into the final whole-genome alignment and visually inspected in Bioedit (Hall 1999).
The resulting alignment was examined for recombination breakpoints using the Genetic Algorithm for Recombination Detection (GARD) method (Pond et al. 2006) and likelihood was evaluated using the Akaike Inference Criterion (AIC). This analysis provided 16 likely breakpoints based on which the whole-genome alignment was split into 17 putatively non-recombinant regions. Phylogenetic reconstruction of each region was performed using RAxML-NG (Kozlov et al. 2019) under a GTR+Γ model. Node support was determined using the Transfer Bootstrap Expectation (TBE) (Lemoine et al. 2018) with 1000 replicates for each tree.
To illustrate the distance of each virus from SARS-CoV-2 while distinguishing whether the virus in question is part of the nCoV clade or the non-nCoV clade, we use an arbitrary tip distance scale normalised between all phylogenies. For each maximum likelihood tree, the tip distance between each tip and SARS-CoV-2 is calculated using ETE 3 for members of the nCoV clade and for members of the non-nCoV clade. These distances are then normalised so that for nCoV clade members they range between 0.1 and 1.1 (1.1 being SARS-CoV-2 itself and 0.1 being the most distant tip from SARS-CoV-2 within the nCoV clade) and between -0.1 and -1.1 for non-nCoV members (-0.1 being the closest non-nCoV virus to SARS-CoV-2 and -1.1 the most distant).
To provide temporal information for the phylogenetic history of the viruses, we performed a Bayesian phylogenetic analysis on non-recombination region 4, using BEAST (Bouckaert et al. 2019). This region was selected due to its length, being the second longest non-recombinant region in the analysis (3764 bp), and the fact that it represents one of the non-recombinant regions where the CoVZC45/CoVZXC21 lineage clusters within the nCoV clade. Based on the observation of an increased evolutionary rate specific to the deepest branch of the nCoV clade reported in MacLean et al. (2020), we adopted the same approach of fitting a separate local clock model to that branch from the rest of the phylogeny. A normal rate distribution with mean 5E-4 and standard deviation 2E-4 was used as an informative prior on all other branches. The lineage containing the BtKY72 and BM48-31 bat viruses was constrained as the outgroup to maintain overall topology. Codon positions were partitioned and a GTR+Γ substitution model was specified independently for each partition. The maximum likelihood phylogeny reconstructed previously for non-recombinant region 4 was used as a starting tree. A constant size coalescent model was used for the tree prior and a lognormal prior with a mean of 6 and standard deviation of 0.5 was specified on the population size. Two independent MCMC runs were performed for 250 million states for the dataset.
References
Boni, MF., P. Lemey, X. Jiang, T. Tsan-Yuk Lam, B.W. Perry, T.A. Castoe, A. Rambaut, and D.L. Robertson. 2020. Evolutionary Origins of the SARS-CoV-2 Sarbecovirus Lineage Responsible for the COVID-19 Pandemic. Nature Microbiology 5 (11): 1408–17.
Bouckaert, R., T.G. Vaughan, J. Barido-Sottani, S. Duchêne, M. Fourment, A. Gavryushkina, J. Heled, et al. 2019. BEAST 2.5: An Advanced Software Platform for Bayesian Evolutionary Analysis. PLoS Computational Biology 15 (4): e1006650.
Gorbalenya, A., S. Baker, R. Baric, R. de Groot, Christian Drosten, A. Gulyaeva, B. Haagmans, et al. 2020. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-nCoV and Naming It SARS-CoV-2. Nature Microbiology 2020: 03–04.
Graham, R.L., and R.S. Baric. 2010. Recombination, Reservoirs, and the Modular Spike: Mechanisms of Coronavirus Cross-Species Transmission. Journal of Virology 84 (7): 3134–46.
Hall, T.A. 1999. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. In Nucleic Acids Symposium Series, 41:95–98.
Hu, D., C. Zhu, L. Ai, T. He, Y. Wang, F. Ye, L. Yang, et al. 2018. Genomic Characterization and Infectivity of a Novel SARS-like Coronavirus in Chinese Bats. Emerging Microbes & Infections 7 (1): 154.
Katoh, K., K.-I. Kuma, H. Toh, and T. Miyata. 2005. MAFFT Version 5: Improvement in Accuracy of Multiple Sequence Alignment. Nucleic Acids Research 33 (2): 511–18.
Kozlov, A.M., D. Darriba, T. Flouri, B. Morel, and A. Stamatakis. 2019. RAxML-NG: A Fast, Scalable and User-Friendly Tool for Maximum Likelihood Phylogenetic Inference. Bioinformatics 35 (21): 4453–55.
Lam, T. Tsan-Yuk, N. Jia, Y.-W. Zhang, M. Ho-Hin Shum, J.-F. Jiang, H.-C. Zhu, Y.-G. Tong, et al. 2020. Identifying SARS-CoV-2-Related Coronaviruses in Malayan Pangolins. Nature 583 (7815): 282–85.
Lemoine, F., J.-B. Domelevo Entfellner, E. Wilkinson, D. Correia, M. Dávila Felipe, T. De Oliveira, and O. Gascuel. 2018. Renewing Felsenstein’s Phylogenetic Bootstrap in the Era of Big Data. Nature. 556: 452–456.
Lin, X.-D., W. Wang, Z.-Y. Hao, Z.-X. Wang, W.-P. Guo, X.-Q. Guan, M.-R. Wang, et al. 2017. Extensive Diversity of Coronaviruses in Bats from China. Virology 507: 1–10.
Liu, P., W. Chen, and J.-P. Chen. 2019. Viral Metagenomics Revealed Sendai Virus and Coronavirus Infection of Malayan Pangolins (Manis Javanica). Viruses 11 (11).
MacLean, O.A., S. Lytras, S. Weaver, J.B. Singer, M.F. Boni, P. Lemey, S.L. Kosakovsky Pond, and D.L. Robertson. 2020. Natural Selection in the Evolution of SARS-CoV-2 in Bats, Not Humans, Created a Highly Capable Human Pathogen. BioXriv https://doi.org/10.1101/2020.05.28.122366.
Pond, S. L. Kosakovsky, S. L. Kosakovsky Pond, D. Posada, M. B. Gravenor, C. H. Woelk, and S. D. W. Frost. 2006. GARD: A Genetic Algorithm for Recombination Detection. Bioinformatics 22 (24): 3096–3098.
Smith, A.T., and Y. Xie. 2013. Mammals of China Edited The Quarterly Review of Biology 88 (4): 363–363.
Suyama, M., D. Torrents, and P. Bork. 2006. PAL2NAL: Robust Conversion of Protein Sequence Alignments into the Corresponding Codon Alignments. Nucleic Acids Research 34: W609–12.
Wang, H., L. Pipes, and R. Nielsen. 2020. Synonymous Mutations and the Molecular Evolution of SARS-Cov-2 Origins. Virus Evolution veaa098, Synonymous mutations and the molecular evolution of SARS-CoV-2 origins | Virus Evolution | Oxford Academic.
Xu, L., J. Guan, W. Lau, and Y. Xiao. 2016. An Overview of Pangolin Trade in China. TRAFFIC September 2016: 1–10.
Zhou, H., X. Chen, T. Hu, J. Li, H. Song, Y. Liu, P. Wang, et al. 2020. A Novel Bat Coronavirus Closely Related to SARS-CoV-2 Contains Natural Insertions at the S1/S2 Cleavage Site of the Spike Protein. Current Biology 30 (11): 2196–2203.e3.
Zhou, P. X.-L. Yang, X.-G. Wang, B. Hu, L. Zhang, W. Zhang, H.-R. Si, et al. 2020. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 579 (7798): 270–73.