Evidence for coordinated evolution at SARS-CoV-2 spike sites that carry Omicron characteristic mutations
A.D. Neverov*1, G.G. Fedonin1,2,3, D.I. Bykova1,4, A.V. Popova1 and G.A. Bazykin5
-
Central Research Institute for Epidemiology, Moscow, Russian Federation
-
Moscow Institute of Physics and Technology (National Research University), Moscow, Russian Federation
-
Institute for Information Transmission Problems of the Russian Academy of Sciences (IITP RAS), Moscow, Russian Federation
-
Moscow State University, Moscow, Russian Federation
-
Skolkovo Institute of Science and Technology, Moscow, Russian Federation
We used a phylogenetic approach to search for coevolving site pairs in the spike protein of SARS-Cov-2. We detected 52 sites which formed 49 coevolving pairs. Sites in coevolving pairs are situated on the protein structure closer to each other than random pairs, and for 41% of these site pairs the residues are in contact. To further characterize this coevolution, we constructed a graph where edges corresponded to coevolving site pairs. 30 of the sites fell into five coevolving clusters, each consisting of between 5 and 9 sites. Three of five connected components form compact clusters on the structure: one is located in the RBD domain near human receptor binding sites, one is located in the flexible part of RBD, and one is in the NTD domain in an area targeted by neutralizing antibodies. Among the 37 sites that carry lineage-defining mutations of the newly identified Omicron variant of concern, at least six show a signal of coevolution with each other.
Introduction
Evolution of SARS-CoV-2 in human hosts before November 2020 was largely neutral, with little evidence for emergence of novel adaptation, and with viral diversity forming by changing periods of globalization and regional diversification [1]. Since the end of 2020, evidence of adaptive viral evolution has started to accumulate, suggesting a change in the mode of evolution [2]. This period was characterized by emergence of multiple concurrently circulating lineages with increased fitness compared to the ancestral variant, including Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1) and Delta (B.1.617.2). At the time of writing, Delta is globally predominant, while a new recently discovered Omicron B.1.1.529 lineage is considered by WHO as the variant of very high risk of global spreading (28 of November Technical Brief and Priority Actions for the Member States). The Omicron variant has a very high number (50) of lineage-defining mutations, most of which (32) are in the S-protein. Interestingly, nine of the sites with lineage defining mutations demonstrated a strong signal of purifying selection against changes of ancestral amino acids before the discovery of Omicron [3]. Purifying selection suggested that Omicron lineage-defining mutations at these sites are individually deleterious. The obvious high fitness of Omicron suggested sign positive epistasis between these sites and/or a possible change of the protein fitness landscape. The Omicron variant had sixteen spike sites that were or had been previously under positive selection, a higher fraction than that observed in spike evolution overall [3].
Several previous studies have attempted to infer the possible epistatic interactions between sites of SARS-CoV-2 genome from sequence data or experimentally. In an early study, Zeng et. al. used direct coupling analysis (DCA) to search for epistasis in the SARS-Cov-2 genome and reported several pairs of putatively interacting sites [4]. No pairwise interactions between sites of the S-protein were identified in this study. In the study of Rochman et al., a method based on counting of mutations on the phylogeny was used to look for strongly associated mutation pairs [1,5]. They found intra- and intergenic epistasis between positively selected mutations in the nuclear localization signal (NLS) of the N-protein and RBD in the S-protein. In RBD, many of the detected epistatically interacting mutations were among the lineage signature mutations. Another proposed approach to study epistatic interaction was to estimate the fitness effects of mutations that arose on the different backgrounds relative to their effects on the wild type background [6]. Using molecular dynamics, Rochman et. al. estimated effects of all individual non-synonymous mutations in the S-protein RBM on binding with host ACE2 receptor and on binding with neutralizing Ab (NAb). Effects of each mutation were estimated for Wuhan ancestral background, Delta (452R, 478K) and Gamma variants (417T, 484K, 501Y). Epistatic effects of mutations weakly stabilized NAb binding for Delta and destabilized it for Gamma variants relative to the ancestral background. Authors concluded that the Gamma variant had more potential for emergence of immune escape mutations than Delta or Wuhan variants.
For some sites, epistasis had been demonstrated experimentally. For example, the Q498R mutation alone affected the affinity of Spike to ACE2 only slightly [7], but together with N501Y, the affinity of binding increased by a factor of four [8].
Here, we study the mutual distribution of spike mutations in the SARS-CoV-2 phylogeny to infer the pairs of sites with evidence of coevolution, as manifested by a propensity of substitutions at these sites to occur rapidly one after the other. We detect 52 such sites combined into 16 coevolving clusters. Many of these sites carry lineage characteristic mutations, including ten sites with Omicron characteristic mutations.
Results and discussion
To obtain a phylogeny representative of SARS-CoV-2 diversity, we downloaded 3 919 063 SARS-Cov-2 complete genome sequences from the GISAID EpiCov™ database available on 07.09.2021 aligned to the WIV04 reference genome. We ignored insertions and deletions relative to the reference sequence, and removed sequences with inframe stop codons in the spike protein. We then clustered the remaining sequences by pairwise distances between S-protein subsequences, allowing three mutations in the S-protein to be the maximal distance within a cluster, resulting in 7348 clusters. For each cluster, we selected one representative sequence of the best quality with the earliest date of sampling. The median date of representative sequences was February 10, 2021. Therefore, the dataset covered approximately equally both characteristic periods of SARS-Cov-2 evolution: the neutral period between Jan-Nov 2020, and the period of antigenic drift between Dec 2020 - May 2021. We classified representative sequences according to pangolin lineages. Most sequences (5721) were of B.1.* sublineages. The variants of concern (VOCs) B.1.1.7 (270 sequences), B.1.351 (127), B.1.617.2 (6) and P.1 (33), as well as variants of note (VONs) A.23.1 (16) and B.1.525 (55), were among representative sequences. The phylogeny of representative sequences was constructed using IQ-TREE [9]. The tree was rooted by the outgroup USA-WA1/2020 (EPI_ISL_404895) that matched the sequence of the putative SARS-Cov-2 progenitor [10,11]. The ancestral sequences at internal tree nodes were reconstructed by TreeTime [12]. We extracted the part of the alignment of complete genome sequences on terminal and internal tree nodes that corresponded to the S gene. Internal branches in the tree without any mutation in S gene were collapsed. The final tree had 1783 internal branches. For each internal branch, we obtained a list of occurred amino acid mutations.
To find coevolving site pairs, we modified our previously developed phylogenetic approach [13–15] to improve the accuracy of detecting concordantly evolving site pairs. Basically, we used as a measure of the concordance of evolution at two sites the epistatic statistic, calculated as the weighted sum of consecutive pairs of mutations at these two sites on the phylogeny, where each mutation pair was taken with exponential penalty for a waiting time for the later mutation [13]. The value of the epistatic statistics for each site pair was calculated similarly to [15], and compared with the null-model distribution of the statistics obtained by simulating independent evolution of sites. For this, we fixed phylogenetic positions of all mutations at the first site of each ordered site pair and permuted phylogenetic positions of mutations at the second site [14]. The power of this method to infer positive and negative epistatic interactions was validated in simulations (upcoming manuscript).
We applied our approach to the distribution of mutations in the S gene on the SARS-Cov-2 phylogeny. We considered all possible unordered pairs of 185 sites with two or more mutations on internal tree branches.
We found 49 concordantly evolved site pairs which comprised 52 sites (under FDR<13%; table 1, figure 1). The distances between coevolving site pairs on the S-protein trimer structure (PDB ID: 7JJJ) were shorter (mean distance – 21.96A) than expected for random site pairs between the 185 sites with two or more mutations on internal branches (mean distance – 35.63A, P=0.001 Mann Whitney test) as well as for random site pairs between the set of 52 own sites (mean distance – 35.75A, P=0.0005 Mann Whitney test). Among the 44 coevolving site pairs for which the distances on the protein structure (PDB ID: 7JJJ) were known, in 18 (41%), the two sites were in contact, i.e., within 5A from each other.
Table 1 . SARS-Cov-2 S-protein coevolving sites. The characteristics of predicted unordered pairs of coevolving sites are shown: site1 and site 2 - coordinates on the S-protein sequence, nominal p-values, the value of epistatic statistics, the total number of consecutive mutation pairs for two corresponding ordered site pairs, numbers of mutations in consecutive pairs in the site1 and in the site2, total numbers of mutations in the site1 and in the site2.
tab1.pdf (243.9 KB)
Figure 1 . The coevolutionary graph.
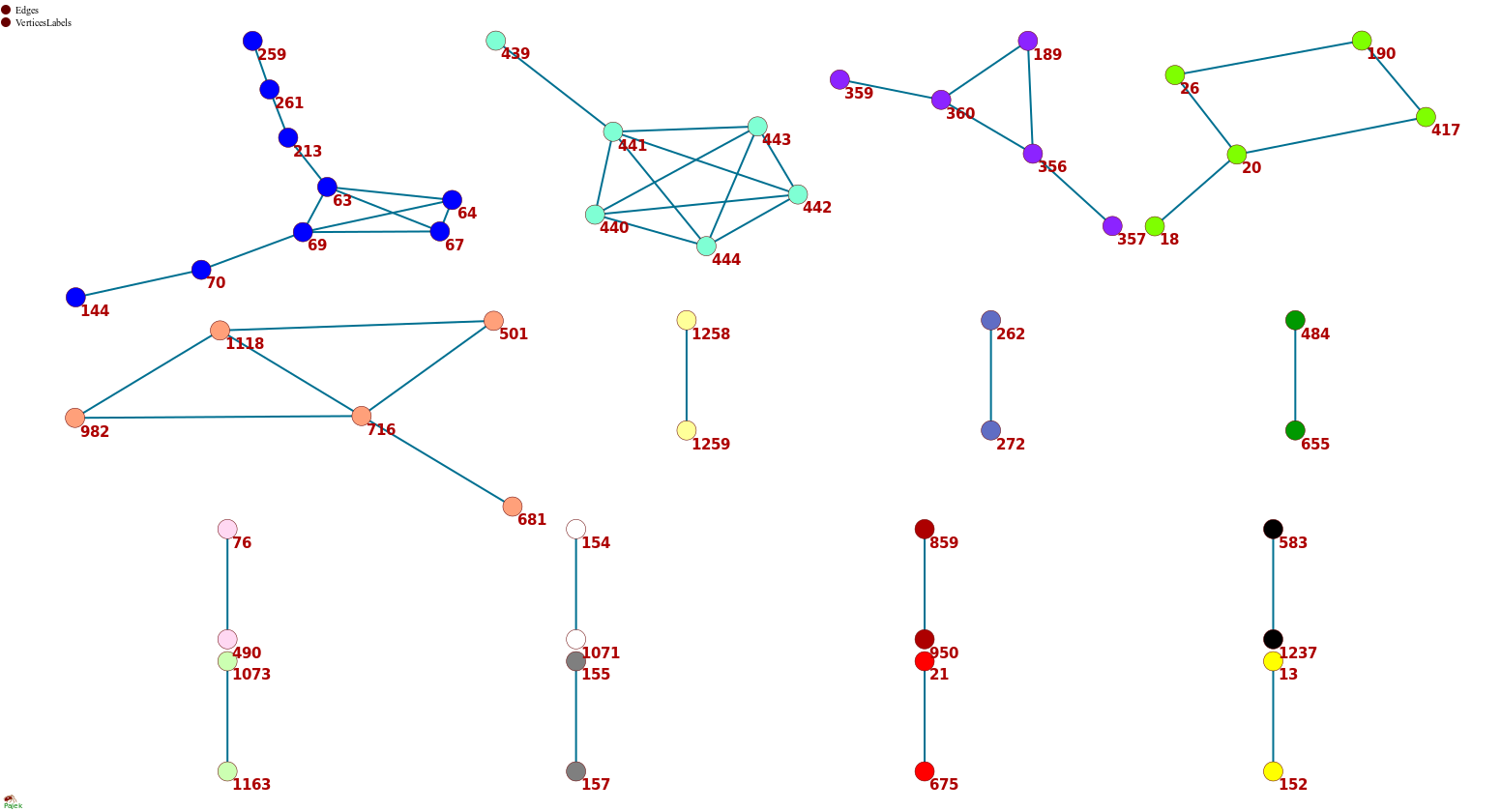
The graph vertices represent sites and edges represent coevolving pairs of sites from the table 1. Graph consists of 16 connecting components, 11 components contain a single edge. Vertices belonging to one connecting component are identically colored.
To get a bird’s eye view on our results, we constructed a coevolutionary graph where vertices represented the 52 sites, and edges represented the 49 pairs (fig. 1). The graph had 16 connected components: five of them were subgraphs each containing between 5–9 sites, and the remaining 11 each consisted of a single site pair. Sites of three of the five subgraphs with multiple vertices formed dense clusters on the protein structure; sites from the other two components were distributed dispersedly (figure 2A). All six sites of the first dense cluster (439-444) were located in the receptor binding motif (RBM) within the receptor binding domain (RBD). The four sites of the second dense cluster (356, 357, 359 and 360) were located in RBD, while the fifth site (189) of the cluster was located in the NTD domain. Interestingly, in the open conformation (PDB ID: 7KL9), sites 357, 359 and 360 contacted with the NTD domain of the adjacent subunit of the S-protein trimer but in the closed conformation (PDB ID: 7jjj) they were not in contacts (fig. 2A and 2B). The third dense cluster (63, 64, 67, 69, 70, 144, 213, 259 and 261) was located in the NTD domain in the region of binding neutralizing antibodies [16]. Four sites (18, 20, 26 and 190) in the first of the two dispersed clusters were located in the NTD domain, and one site (417) was in the RDB. The second dispersed cluster comprised sites from different domains: RBM (501), near S1/S2 cleavage site (681), S2 (716, 982) and in the connecting domain (CD) (1118).
Figure 2 . The clusters of coevolving sites on the protein structure.
Sites of five connecting components of the coevolutionary graph that comprise multiple coevolving pairs of sites are shown. The coevolving sites are shown as spheres, sites of the same cluster are identically colored. The colors correspond to colors of sites on the figure 1. A. The closed conformation of S-protein trimer (pdb: 7JJJ). B. Open conformation of S-protein trimer (pdb: 7KL9): for clarity, only residues 320-590 of one subunit and NTD (14-303) of the adjacent subunit are shown. NTD is shown in dark gray; residues 357, 359 and 360 are shown in dark purple.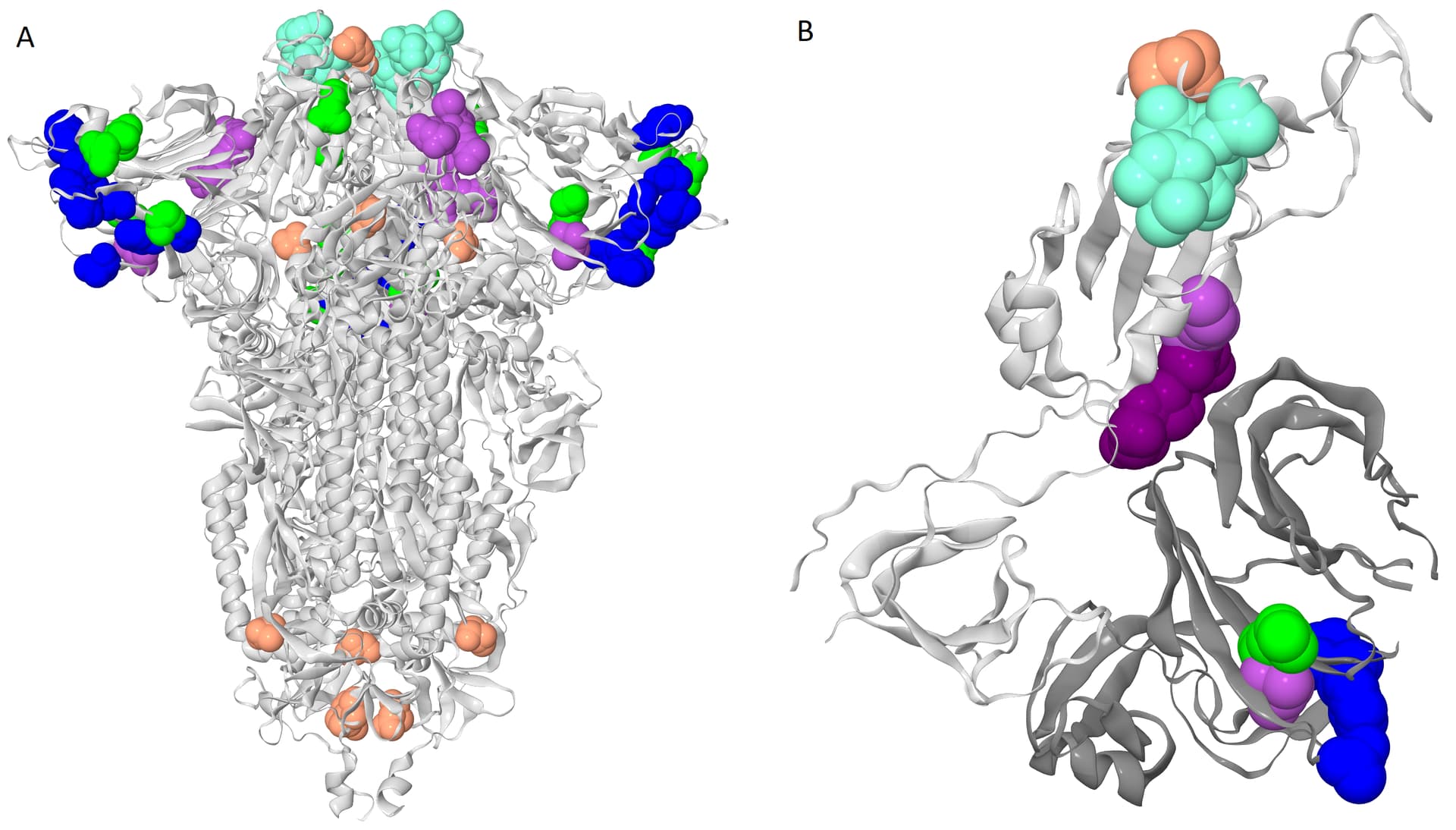
Many of the coevolving sites carried mutations that defined VOC lineages. In what follows, we refer to sites bearing lineage-defining mutations as lineage defining sites.
For the Alpha VOC (B.1.1.7), eight (69, 70, 144, 501, 681, 716, 982 и 1118) out of ten lineage defining sites were among the 52 coevolving sites. Three sites, 69, 70 и 144, were within the third dense cluster of sites located in the NTD; the remaining five sites represented the second dispersed cluster of sites.
For the Beta VOC (B.1.351) , three sites (417, 501 and 484) out of six were among the set of coevolving sites, but these sites had no coevolving partners among the sites with lineage-defining mutations.
For the Gamma VOC (P.1), seven out of ten sites (18, 20, 26, 417, 484, 501 and 655) were among the set of coevolving sites. Four sites (18, 20, 26 and 417) were within the first dispersed cluster of sites. Two sites constituted a separate cluster with a single coevolving pair (484, 655) (figure 3). The site 501 had no coevolving partners.
Figure 3 . Coevolution of S-protein sites 484 and 655.
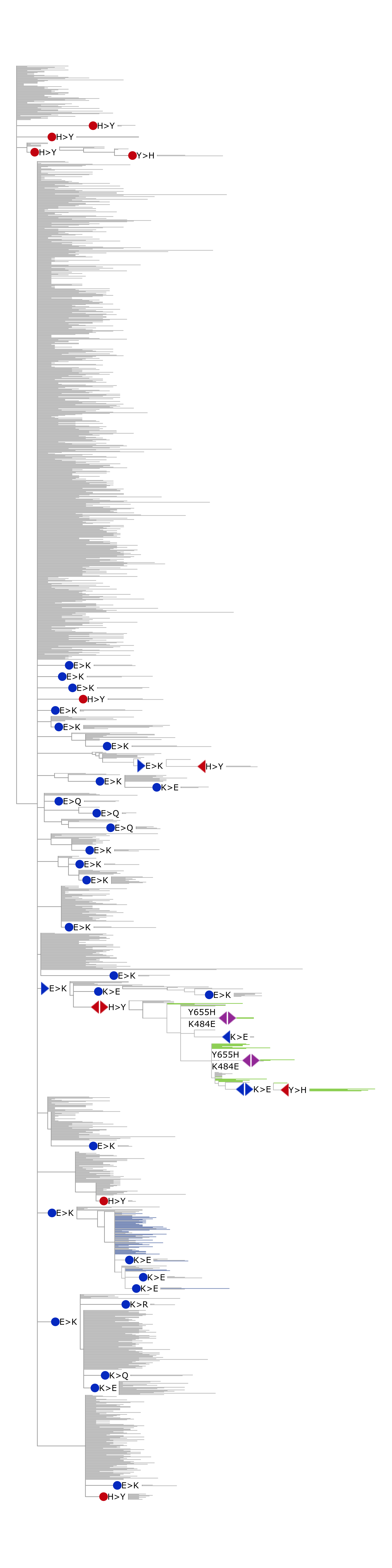
Mutations in two sites that occurred on internal branches of the phylogenetic tree are shown. For illustrative purposes, some clades without mutations in these sites were cut out. Mutations in the sites are shown as marks of blue for the 484 site and red for the 655 site colors. Leading mutations occurring first in corresponding consecutive mutation pairs are shown as right arrows, trailing mutations occurring last are shown as left arrows. Mutations occurring in both sites on the same branches are shown as double violet arrows. Mutations that are not descended to any mutation in another site as well as which are not ancestors for mutations in another site are shown as circles. The green branches correspond to the sequences of Gamma VOC.
For the Delta VOC (B.1.617.2+AY.*) , three out of eight sites (157, 681 and 950) were among the set of coevolving sites, however, these sites belonged to different clusters and they had no coevolving partners among the lineage-defining sites in the S-protein.
Finally, for the Omicron VOC (B.1.529; B.1.1 decendant associated with Southern Africa with high number of Spike mutations · Issue #343 · cov-lineages/pango-designation · GitHub) ten sites (67, 69, 70, 144, 417, 440, 484, 501, 655 and 681) were among the set of coevolving sites. Four sites (67, 69, 70 and 144) were within the third dense cluster of sites located in the NTD. Pair of coevolving sites 501 and 681 belonged to the second highly dispersed cluster of sites but they didn’t coevolve directly. The first site in the pair was located within RBM and the second was located close to S1/S2 furin cleavage site (FS). Pair of coevolving sites 484 and 655 represented a separate one-edge cluster (figure 1 and figure 3). Again, the first site in this pair was within the RBM and the second site was within FS. However, allele 484A that is inherent for the Omicron VOC is likely deleterious for previously circulated strains: mutation E484A occurred on a few (8 out of 7348) terminal branches of our phylogenetic tree and always in the context of ancestral allele histidine in the site 655. The pair of alleles 484A and 655Y was observed in 14 out of the 3 919 063 isolates in GISAID and all of these 14 isolates belong to the Delta VOC, to the seven different complete genome genotypes and to the two different S-gene genotypes. Mutations E484A and H655Y are significantly associated with each other among different S-gene genotypes of Delta VOC (P=0.004 Fisher’s exact test; observed 2 double-mutation genotypes, whereas expected number of genotypes is 0.1). So, the joint preference of E484A and H655Y is likely conditioned on alleles in other sites.
Two sites 440 and 417 had no coevolving partners among the lineage-defining sites in the S-protein. Seven of ten coevolving sites, with exception of sites carrying deletions in Omicron (69, 70 and 144), previously were shown as sites evolving under positive selection [3].
Our findings show that for three out of five VOCs, Alpha, Gamma and Omicron, a fraction of the lineage-defining mutations are found at coevolving pairs of sites. The signal of coevolution for these sites we observed in the dataset which contained the last sample dated by May of 2021 and thus it didn’t include the Omicron sequences.
The observed signal of coevolution could come from at least two sources. First, it could arise from epistatic interactions between these sites, a possibility which we favored in our previous work [13–15]. Second, it could be observed if some fraction of evolutionary time is spent by SARS-CoV-2 in a distinct selection regime. It has been hypothesized that such a selection regime could characterize the origin of at least some of the VOCs, perhaps due to their origin in immunocompromised individuals or in a reverse zoonosis event [3]. Direct experimental studies of the possible epistatic interactions between coevolving sites will help distinguish between these two hypotheses.
Assuming that the positive epistasis is the origin of observed site coevolution, we can conclude that reversions of mutations in sites would be deleterious if there are mutations in the coevolving partner sites. Mutations N440K and K417N have no mutations in partner sites in Omicron. Their population frequencies within the BA.1 sublineage fluctuate around 0.6 since the beginning of November in Africa and steadily decrease in Europe (figure 4). It was shown that these mutations allow to escape of some neutralizing antibodies, so that could be beneficial [17], but their population frequencies could still decrease if trade-offs with other effects of these mutations on viral fitness are negative. If appropriate mutations in the partner sites will arise in future, e.g. in the site 190, these mutations could become fixed. Some mutations presenting now in population of Omicron strains with low frequencies T716I and E484K could be beneficial and they could increase their frequencies because there exist high frequency mutations in their partner sites: N501Y and P681H for T716I and H655Y for E484K.
Figure 4 . Population frequencies of Omicron S-gene mutations K417N (A) and N440K (B).
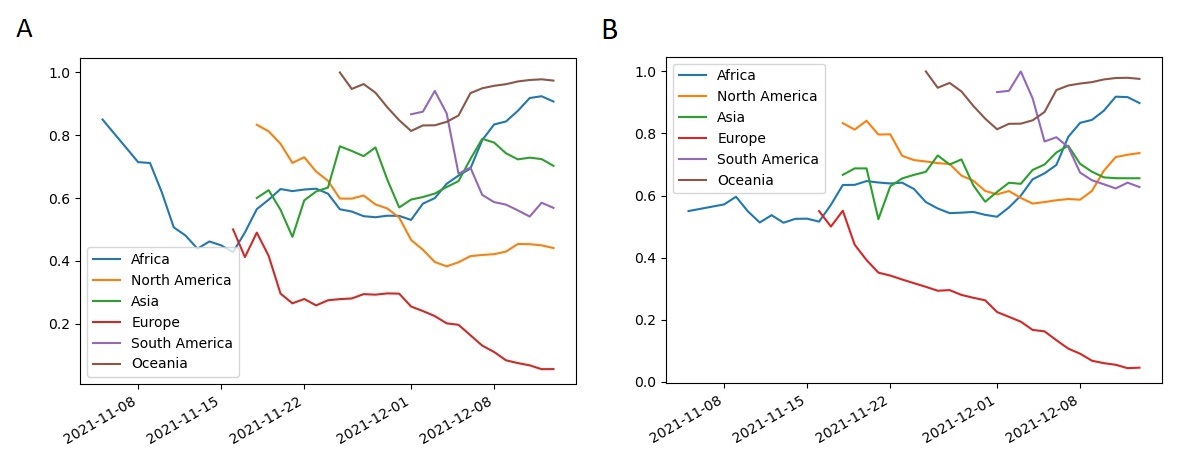
The frequencies of mutations are shown for the dominating sublineage BA.1 for different regions of the World. The mutation counts were aggregated over a 7 days moving window. The points with less than ten samples were not shown.
Literature
-
Rochman ND, Wolf YI, Faure G, et al. Ongoing global and regional adaptive evolution of SARS-CoV-2. Proc. Natl. Acad. Sci. 2021; 118:
-
Martin DP, Weaver S, Tegally H, et al. The emergence and ongoing convergent evolution of the N501Y lineages coincides with a major global shift in the SARS-CoV-2 selective landscape. MedRxiv Prepr. Serv. Health Sci. 2021; 2021.02.23.21252268
-
Martin et al. Selection analysis identifies significant mutational changes in Omicron that are likely to influence both antibody neutralization and Spike function (Part 1 of 2) - SARS-CoV-2 coronavirus. Virological 2021;
-
Zeng H-L, Dichio V, Horta ER, et al. Global analysis of more than 50,000 SARS-CoV-2 genomes reveals epistasis between eight viral genes. Proc. Natl. Acad. Sci. 2020; 117:31519–31526
-
Rochman ND, Wolf YI, Koonin EV. Deep phylogeny of cancer drivers and compensatory mutations. Commun. Biol. 2020; 3:1–11
-
Rochman ND, Faure G, Wolf YI, et al. Epistasis at the SARS-CoV-2 RBD Interface and the Propitiously Boring Implications for Vaccine Escape. 2021; 2021.08.30.458225
-
Starr TN, Greaney AJ, Hilton SK, et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020; 182:1295-1310.e20
-
Zahradník J, Marciano S, Shemesh M, et al. SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat. Microbiol. 2021; 6:1188–1198
-
Minh BQ, Schmidt HA, Chernomor O, et al. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020; 37:1530–1534
-
Bloom JD. Recovery of Deleted Deep Sequencing Data Sheds More Light on the Early Wuhan SARS-CoV-2 Epidemic. Mol. Biol. Evol. 2021; 38:5211–5224
-
Kumar S, Tao Q, Weaver S, et al. An Evolutionary Portrait of the Progenitor SARS-CoV-2 and Its Dominant Offshoots in COVID-19 Pandemic. Mol. Biol. Evol. 2021; 38:3046–3059
-
Sagulenko P, Puller V, Neher RA. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018; 4:vex042
-
Kryazhimskiy S, Dushoff J, Bazykin GA, et al. Prevalence of Epistasis in the Evolution of Influenza A Surface Proteins. PLOS Genet. 2011; 7:e1001301
-
Neverov AD, Kryazhimskiy S, Plotkin JB, et al. Coordinated Evolution of Influenza A Surface Proteins. PLOS Genet. 2015; 11:e1005404
-
Neverov AD, Popova AV, Fedonin GG, et al. Episodic evolution of coadapted sets of amino acid sites in mitochondrial proteins. PLOS Genet. 2021; 17:e1008711
-
McCarthy KR, Rennick LJ, Nambulli S, et al. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science 2021; 371:1139–1142
-
Cao Y, Wang J, Jian F, et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2021;