Emergence of Y453F and Δ69-70HV mutations in a lymphoma patient with long-term COVID-19
Bazykin GA1,2*, Stanevich O3,4*, Danilenko D4, Fadeev A4, Komissarova K4, Ivanova A4,
Sergeeva M4, Safina K1,2, Nabieva E1, Klink G2, Garushyants S2, Zabutova J5, Kholodnaia A3,5, Skorokhod I5, Ryabchikova VV5, Komissarov A4, Lioznov D3,4
1 Skolkovo Institute of Science and Technology (Skoltech), Moscow, Russia
2 A.A. Kharkevich Institute for Information Transmission Problems of the Russian Academy of
Sciences, Moscow, Russia
3 First Pavlov State Medical University, Saint-Petersburg, Russia
4 Smorodintsev Research Institute of Influenza, Saint-Petersburg, Russia
5 City Hospital 31, Saint-Petersburg, Russia
*equal contribution; [email protected]
Abstract
We report a genomic analysis of SARS-CoV-2 from a lymphoma patient with long-term COVID-19. This genome is characterized by an independent gain of 18 new mutations over more than 4 months of the disease. These include the S:Y453F and Δ69-70HV mutations (“the ΔF combination”) which have formerly been associated with mink-related clusters. Both of these mutations are found at intermediate frequencies in the patient, representing a case of intra-host polymorphism. Phylogenetic analysis indicates that the patient’s lineage is not related to the mink cluster, indicating that these mutations were gained anew. Independent acquisition of an identical pair of mutations in a mink and a lymphoma patient, and between multiple immunosuppressed patients, suggests concordant changes in selection.
Introduction
Persistently positive PCR test for SARS-CoV-2 occurs in a fraction of COVID-19 patients, and is usually associated with suppressed host immune system1–4. In these individuals, SARS-CoV-2 seems to undergo rapid accumulation of mutations, as evidenced by long phylogenetic branches5,6. The question remains to what extent persistent RNA positivity can drive adaptive evolution of SARS-CoV-2 at population level7.
Spread of SARS-CoV-2 in minks caused global concern, leading to culling of millions of minks in Denmark. Reasons for alarm included recurrent transmission into minks, rapid spread of the virus in minks, detection of a mink-specific set of mutations at functionally important sites, some of which caused immune escape and/or affected ACE2 binding, and transmission back to humans8–10.
The spillover of SARS-CoV-2 into mink populations has been associated with recurrent acquisition of a number of mutations in the spike protein, which were preserved in subsequent mink-to-human transmission. While a total of 5 mink-associated mutations were identified, a combination of just two of them, Δ69-70HV + Y453F (a.k.a. ΔF), is responsible for the majority of positive samples10. The ΔF combination confers ability to rapidly replicate to high titers and to potentially evade recognition by neutralizing antibodies10, raising concerns that these mutations may affect vaccine efficiency. Y453F affects the receptor-binding domain (RBD), possibly increasing hACE2 binding11,12. It allows immune escape from monoclonal antibodies and polyclonal sera12; in particular, it has led to 57% escape from the REGN10933 monoclonal antibody, a component of FDA-approved Regeneron’s REGN-COV2 cocktail for treatment of COVID-19 patients, although it did not allow escape from the full cocktail of two antibodies (REGN10933+REGN10987)13. Δ69-70HV has arisen repeatedly14, and is involved in evasion of neutralizing antibodies5. The ΔF combination of mutations has arisen in parallel in multiple mink populations; among humans, it was mainly found in cases traceable to minks, indicating reverse transmission15.
Here, we report a case of a lymphoma patient with a long-term COVID-19, with positive PCR tests spanning a period of over 4 months. We show that she has acquired 18 mutations de novo, including the ΔF combination.
Results
Detailed case history will be presented elsewhere. In brief, patient S, female, aged 47, diagnosed with non-Hodgkin diffuse B-cell lymphoma IV stage B, was admitted to the hospital for planned chemotherapy on March 27 and was discharged on April 17, 2020. In the period between April 5 - April 8, she received chemotherapy with monoclonal antibody rituximab, R-ICE regimen. Between April 10 and April 16, she had close contacts with an elderly woman, patient A, who was transferred to her ward and later was tested positive by PCR for COVID-19 (the swab was taken on April 10). Patient A later died of COVID-19 pneumonia; paraffin blocks with post-mortem material from her were subsequently analyzed for SARS-CoV-2 by PCR, followed by RNA extraction and sequencing.On April 17, 2020, patient S was discharged from the hospital. On April 30, a nasopharyngeal swab for SARS-CoV-2 performed by an outpatient doctor was positive. By that time, the patient had an onset of COVID-19 symptoms (subfebrile temperature). That original sample was discarded directly after testing and was not available for sequencing. Repeated swabs from May 14, May 19, June 9 and July 14 were SARS-CoV-2 negative. Nevertheless, patient S demonstrated symptoms of severe COVID-19 between May 25 - August 21. She tested positive again on August 03, August 5, August 8, August 11, August 13, August 17, August 20, August 21, August 26, August 27, September 03 and September 9; she finally tested negative on September 12, 2020, and again on November 10 and December 16.
Next-generation sequencing was performed on a nasopharyngeal swab sample obtained from patient S from the relapse dating to 20.08 (hCoV-19/Russia/SPE-RII-30769S/2020). To root this lineage, we also sequenced a sample from swab of patient A, the probable source of infection, obtained on April 10.
25 genetic changes distinguish the patient S sample from the Wuhan-Hu-1/2019 reference strain (Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, co - Nucleotide - NCBI). Of them, seven SNPs, including the three SNPs at adjacent positions 21881-21883, place the patient’s sample in the B.1.1 lineage. The lineage of patient S carries the remaining 18 genetic changes. The patient A sample is positioned at the root of the B.1.1 lineage, confirming that the 18 mutations are specific to patient S (Fig. 1).

Fig. 1. Position of patient S in the B.1.1 lineage. The phylogenetic tree of B.1.1 reconstructed for 49,083 sequences was pruned to contain a random set of 1% of all samples, including the sample from patient S (red dot). Patient A (black dot) matches the ancestral state of the B.1.1 lineage. The lineage of patient S (red) totals 18 mutations. These include S:Δ69-70HV and S:Y435F, marked in the two inner circles in yellow and blue respectively. The B.1.1.7 lineage and cluster 5 are shaded.
The PANGOLIN package assigns the sample of patient S to the B.1.1.163 lineage on the basis of the A12886G mutation. Although this lineage is predominantly composed of Russian samples, this placement is inconsistent with the phylogenetic position of the patient A sample, which roots the patient S sample at the base of the B.1.1 lineage. Such discrepancy can be explained by the presence of private mutations in the patient S sample which support conflicting positions of the sample within the B.1.1 lineage (Fig. S1). In any case, patient S cannot be placed into the cluster 5 clade because cluster 5 is separated from B.1.1 by two additional mutations (those at positions 15656 and 25936) which are absent in patient S (Fig. S1).
Among the 18 lineage-specific patient S mutations, 3 are deletions, including S:Δ69-70HV. Another observed deletion is S:Δ141-144; the same deletion has also originated previously in another immunocompomised patient1. The remaining 15 changes are single-nucleotide mutations; of them, 10 are missense (amino acid-changing), 1 is nonsense (stop codon-creating) (Table 1), and the remaining 4 are silent (amino acid-preserving). The estimated ratio of the number of nonsynonymous and synonymous substitutions for this branch is 0.99; this is higher than the mean across all external lineages leading to samples of the B.1.1 clade (mean = 0.57, median = 0.55), although the difference is not significant (p = 0.1). Among the 15 nonsynonymous mutations, 5 (33.3%) occur in the spike protein which accounts for 13% of the genome, and two (13%), Y453F and T470N, occur in the RBD which accounts for 2% of the genome. The observed nonsense mutation occurred in codon 18 of ORF8.
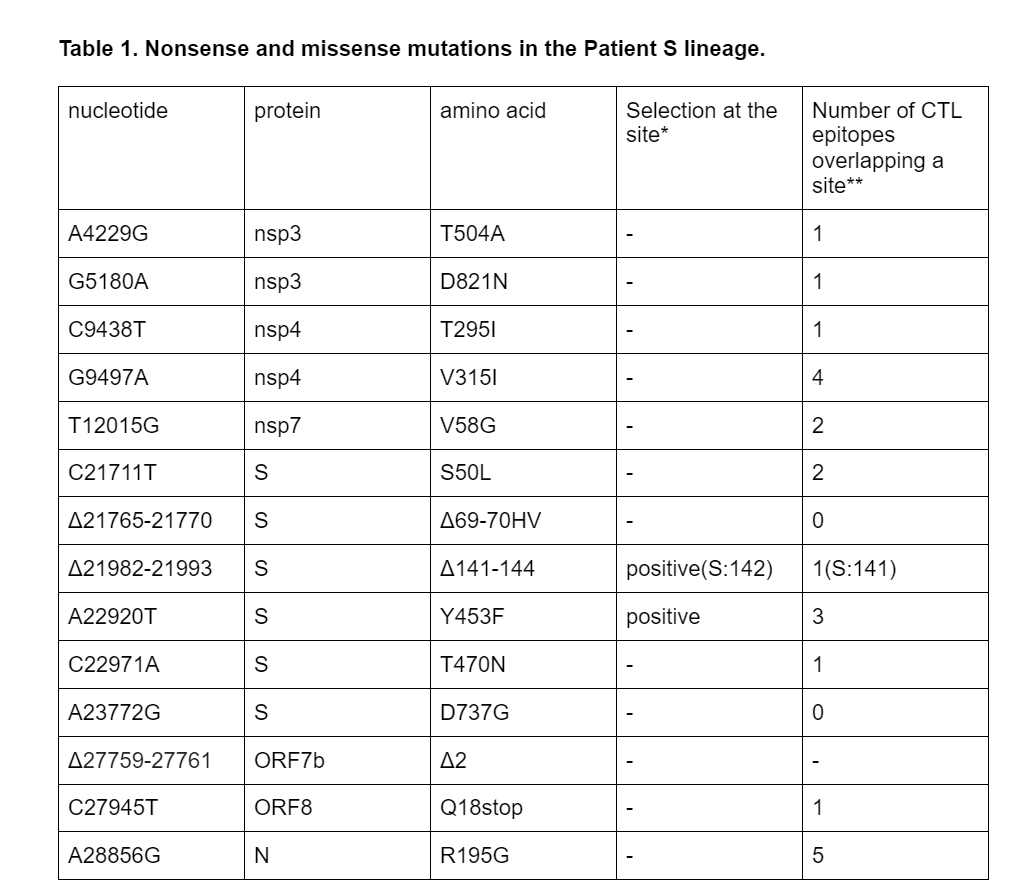
*according to ref. 16, with significance cut-off 0.05
**according to ref. 17
To validate the phylogenetic position of patient S samples, we sequenced two additional samples from the relapse, dating to 17.08 and 20.08. The three samples from the two timepoints matched at all SNPs that were called in the consensus sequence, and carried within-patient polymorphism at the same positions (Fig. 2).
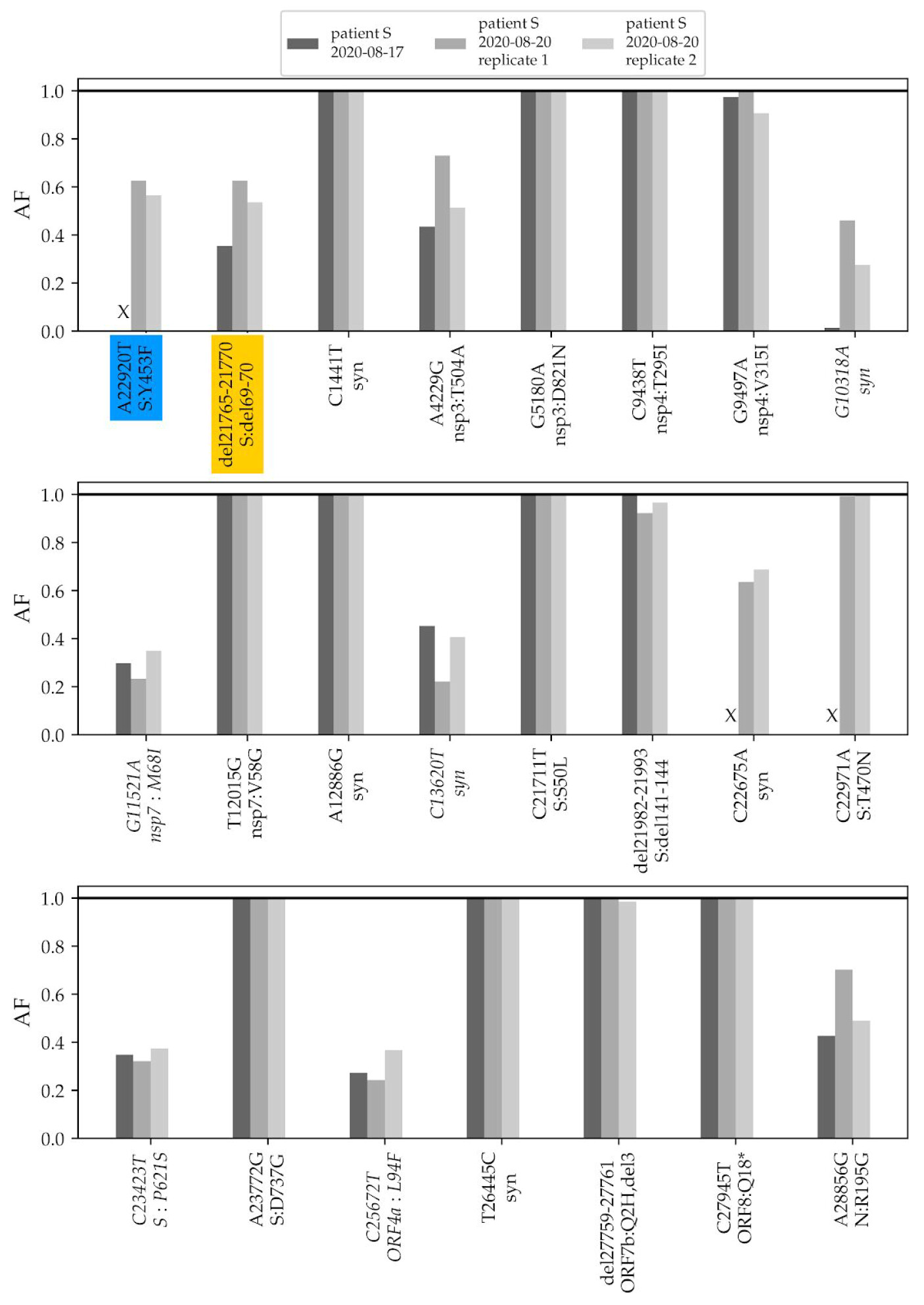
Fig. 2. Frequencies of mutations of the patient S lineage in her three samples. The first two variants (highlighted in color) are shared with cluster 5. The remaining variants (ordered by genomic coordinate) are specific to the lineage of patient S (the seven mutations that place the samples to the B.1.1 lineage are omitted). “X” represents absence of coverage at a position (<4 reads). Only variants that reach at least 30% frequency in any of the three samples are shown; variants that do not reach 50% in any of the samples are italicized. AF, allele frequency; Syn, synonymous (silent) variants.
The changes in the lineage leading to the patient include the ΔF combination. A closer examination indicates that these mutations were polymorphic within the samples from both timepoints. The Δ69-70HV mutation was observed in 28% of the reads in the August 17 sample, and in 63% and 54% of the reads in the two August 20 samples (Fig. 2). The Y453F mutation was observed in 63% and 56% of the reads in the 20.08 sample (in the 17.08 sample, this position was insufficiently covered).
In addition to these two variants, patient S carried another 8 polymorphic mutations at substantial frequencies (above 30% in at least one of the samples; Fig. 2). Among the two remaining cluster 5 mutations, one, I692V, was supported by 8 out of the 1246 sequencing reads (0.6%) in one of the samples (3/1875 and 2/376 in the two others), likely representing sequencing errors; the other one, M1229I, was not observed.
Discussion
The SARS-CoV-2 lineage of patient S underwent rapid evolution within the host. The observed 18 changes accumulated between April 10 and August 20, i.e. over the course of at most 132 days, correspond to a rate of 1.67E-3 changes/nucleotide/year, substantially exceeding the average rate of evolution of SARS-CoV-2. Accelerated evolution of SARS-CoV-2 in an immunocompromised patient is in line with previous findings1,5,6. The observed excess of nonsynonymous changes suggests that this rate increase is caused by relaxed selective constraint and/or positive selection, rather than an increase in the overall mutation rate or genome editing. The observed excess of changes in the spike protein, and in particular in the RBD, is consistent with the effect of these mutations on hACE2 binding and/or antibody avoidance; in addition, the abundance of nonsynonymous changes at other proteins may suggest a role of cytotoxic T-lymphocyte escape. Some of the acquired variants occur at intermediate frequencies, representing within-host polymorphism.
As of today, patient S represents the only sample in GISAID with the combination of S:Y453F and S:Δ69-70HV mutations outside of cluster 5, i.e., acquired independently of minks (Fig. 3). The genomic signature of cluster 5 has been used to confirm that farm workers have been infected from the animals 9; however, we now show that getting two of these four mutations is generally not sufficient to call such directionality. While cluster 5 is now probably extinct18, our observation of independent acquisition of two of its mutations, both with likely functional effect, in a human may point to the risk of their subsequent onward transmission.
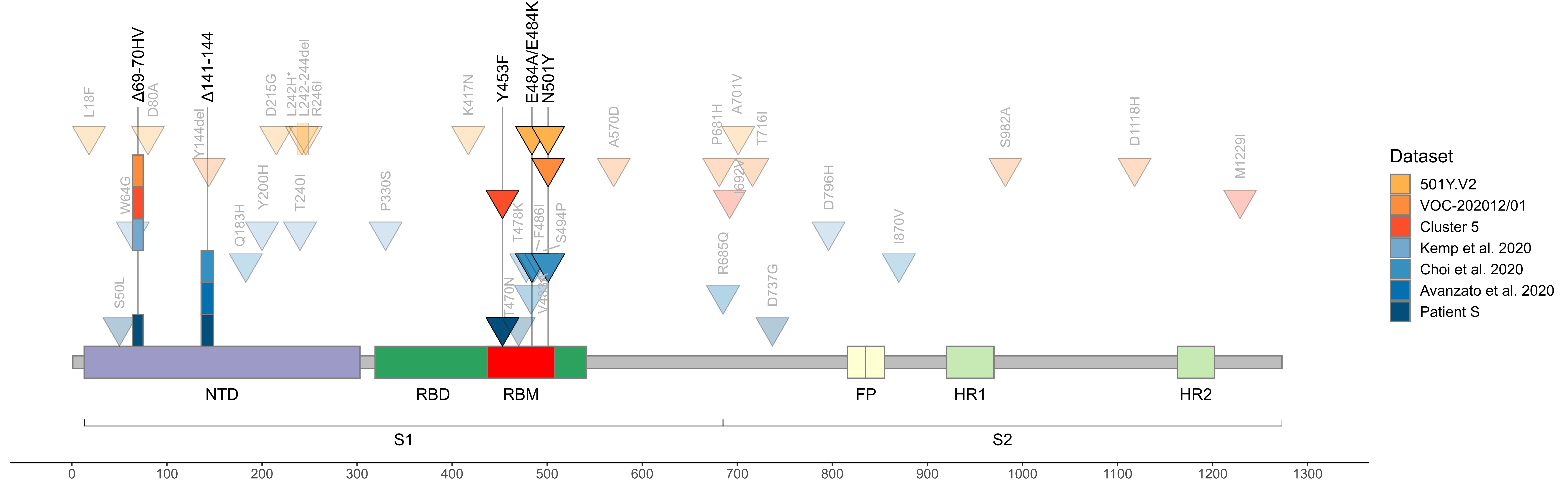
Fig. 3. Concordant origin of spike mutations in notable COVID-19 variants and reported cases of persistent COVID-19. Shown are the locations of mutations in the amino acid sequence encoded by the spike gene. Rows, from top to bottom: 501Y:V2 variant; VOC-202012/01 variant; cluster 5 variant; immunosuppressed individual treated with convalescent plasma (Kemp et al. 2020, ref. 5); immunosuppressed individual treated with Regeneron monoclonal antibody cocktail (Choi et al. 2020, ref. 6; only those mutations present at the final timepoint (T3, day 152) are shown); immunocompromised individual treated with convalescent plasma (Avanzato et al. 2020, ref. 1); immunosuppressed individual not treated with convalescent plasma or antibodies (patient S, this study). Triangles, point mutations; rectangles, deletions. Bright colors represent mutations observed in at least two studies.
Outside minks, S:Δ69-70HV was recently shown to occur in a virus from another immunocompromised patient with COVID-195. In that study, S:Δ69-70HV has been fixed during convalescent plasma therapy, suggesting antibody selection pressure, which is consistent with decreased virus sensitivity to neutralisation with sera from recovered patients. However, patient S was not treated with convalescent plasma, was taking rituximab, the B-cell-depleting agent, and had no detectable neutralizing antibody response. This suggests that this mutation could have been favored by some other factor of selection. In patient S, both the S:Y453F and the S:Δ69-70HV mutations remain polymorphic, indicating that they were not yet fixed by selection in their favor; alternatively, the presence of both the ancestral and the derived variant at these sites could be maintained by balancing selection, although it is impossible to test this hypothesis with just one time point.
In addition to S:Y453F and S:Δ69-70HV, patient S has acquired two other mutations that were also preferentially obtained by other immunocompromised patients. The first is S:Δ141-144, which has previously arisen in another immunocompromised patient in a case of persistent COVID-191 (Fig. 3). The second is an early termination of translation (nonsense mutation) in ORF8, which has occurred in the 18th codon in patient S. While the functions of ORF8 and its role in disease are extensively debated19,20, a different nonsense mutation in the 27th codon of ORF8 is one of the lineage-defining mutations of cluster B.1.1.7 which is thought to be founded by a chronically infected individual7. Given that ORF8 is thought to suppress immune response, selection against its loss may be relaxed in immuno-compromised patients. Of note, the S:N501Y mutation which currently rapidly spreads as part of the B.1.1.7 and 501.V2 clades was also previously found in a longitudinal study of an immunocompromised patient where it has been acquired after ~3 months of infection6.
The fact that all these mutations are acquired concordantly (Fig. 3) indicates that they are favored by selection pressure which is common to immunocompromised patients, and possibly to minks, but distinct from that in the general population. Nevertheless, the variants acquired in immunocompromised patients (and in minks) can later spill over into the general population, affecting the characteristics of circulating strains.
Methods
Consensus calling
Raw reads were trimmed with Trimmomatic-0.39 (http://www.usadellab.org/cms/?page=trimmomatic) to remove adapter sequences and low-quality ends. Trimmed reads were mapped onto the Wuhan-Hu-1 (MN908947.3) reference genome with bwa mem. The following reads were then removed from the mapping: reads with abnormal insert length to read ratio (greater than 10 or less than 0.8), reads with insert length greater than 1100, reads with more than 50% soft-clipped bases. Soft-clipped ends were trimmed from the remaining reads, 10 nucleotides were cropped from read ends using custom scripts, and primer sequences were removed with ivar (https://andersen-lab.github.io/ivar/). Only reads with at least 30 nucleotides remaining after the procedure were kept. SNV and short indel calling was done with lofreq (https://csb5.github.io/lofreq/), with SNVs considered consensus if they were covered by at least 4 reads and supported by more than 50% of those reads; indels were considered consensus if they were covered by at least 20 reads with at least 50% of those supporting the variant. Regions that were covered by fewer than 2 reads or that were covered by 2 or 3 reads and called non-reference were masked as N. Consensus was created by bcftools consensus (http://samtools.github.io/bcftools/bcftools.html).Bioinformatics analysis
255,389 genomes of SARS-CoV-2 were downloaded from GISAID on December 12, 2020, (Supplementary Data ACKN) and aligned with MAFFT v7.453 71 against the reference genome Wuhan-Hu-1/2019 (NCBI ID: MN908947.3) with --addfragments --keeplength options. 100 nucleotides from the beginning and from the end of the alignment were trimmed. After that, we excluded sequences (1) shorter than 29,000 bp, (2) with more than 300 positions of missing data (Ns) and gaps, (3) excluded by Nextstrain, (4) from animals other than minks, or (5) corresponding to resequencing of the same patients, leaving 201,948 sequences. Identical sequences were then collapsed within each country and host and annotated by the Pangolin package. As the sequence of patient S (EPI_ISL_596228) from the current study is annotated as B.1.1.163 lineage by Pangolin, we further only kept sequences annotated as B.1.1, excluding a large clade defined by mutation G25563T (GH clade in GISAID nomenclature). We additionally masked a highly-homoplasic site 11083. The final set of 49,083 sequences was used to construct the phylogenetic tree with IQ-Tree v2.1.1 72 under GTR substitution model and ‘-fast’ option. Ancestral sequences at the internal tree nodes were reconstructed with TreeTime v0.8.0 73. Pairwise dN/dS between every sample (terminal node) and its nearest ancestor was calculated with codeml program from PAML package (version 4.6) (https://academic.oup.com/mbe/article/24/8/1586/1103731?login=true). To produce the distribution of dN/dS ratios, samples with dN = 0 or dS = 0 were excluded. P-value for Patient S was calculated as the fraction of samples with dN/dS more than or equal to that for Patient S.Supplementary Figure
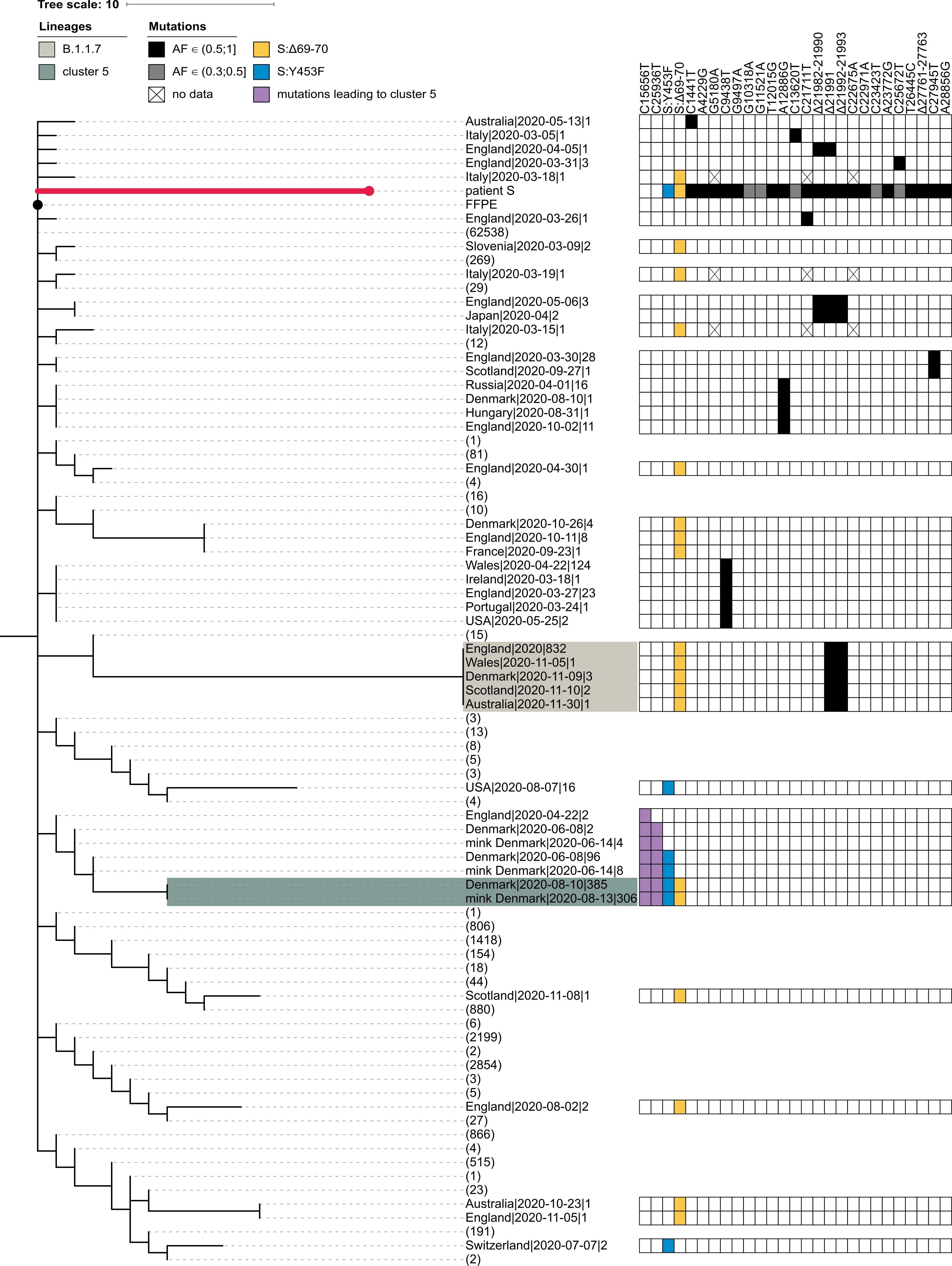
Fig. S1. Patient S cannot be placed in the cluster 5 clade. The abridged phylogeny of the B.1.1 lineage phylogeny is shown. Only those samples are shown which met either of the following conditions: (i) it carried any of the differences found between the B.1.1 root and the patient S sample (black cells), and these mutations had occurred in the branch immediately descendant from the B.1.1 root; or (ii) it carried either the S:Δ69-70HV (blue cells) or the S:Y435F (yellow cells) mutation, independent of the timing of their origin. Additionally, we retained the samples from the branches that separate the cluster 5 clade from the rest of the phylogeny (two additional mutations, purple cells). Samples that didn’t meet these criteria were collapsed, with the number of such samples shown in parentheses. The retained samples were then grouped by country, with names formatted as ‘country|date of the earliest sample|number of samples’. B.1.1.7 and cluster 5 samples are shaded as in Fig. 1. The presence of the above-mentioned mutations is indicated by the matrix at the right. Two mutations distinguishing cluster 5 from the B.1.1 root (purple) reject uniting patient S and cluster 5 in the same clade. For patient S, mutations with allele frequency below 50% in all three samples are shown in grey. Missing data (‘N’s in sequences) are shown as crosses. FFPE, patient A (the presumed source of infection for patient S).
References
1 Avanzato VA, Matson MJ, Seifert SN, et al. Case Study: Prolonged Infectious SARS-CoV-2 Shedding from an Asymptomatic Immunocompromised Individual with Cancer. Cell 2020; 183: 1901-1912.e9.
2 Karataş A, İnkaya AÇ, Demiroğlu H, et al. Prolonged viral shedding in a lymphoma patient with COVID-19 infection receiving convalescent plasma. Transfus Apher Sci 2020; 59. DOI:10.1016/j.transci.2020.102871.
3 Nakajima Y, Ogai A, Furukawa K, et al. Prolonged viral shedding of SARS-CoV-2 in an immunocompromised patient. J Infect Chemother 2020; 0. DOI:10.1016/j.jiac.2020.12.001.
4 Wei L, Liu B, Zhao Y, Chen Z. Prolonged shedding of SARS-CoV-2 in an elderly liver transplant patient infected by COVID-19: a case report. Ann Palliat Med 2020; 0. DOI:10.21037/apm-20-996.
5 Kemp SA, Collier DA, Datir R, et al. Neutralising antibodies drive Spike mediated SARS-CoV-2 evasion. Infectious Diseases (except HIV/AIDS), 2020 DOI:10.1101/2020.12.05.20241927.
6 Choi B, Choudhary MC, Regan J, et al. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. N Engl J Med 2020; 383: 2291–3.
7 Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. Virological. 2020; published online Dec 18. https://pando.tools/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563/1 (accessed Dec 30, 2020).
8 Rapid Risk Assessment: Detection of new SARS-CoV-2 variants related to mink. Eur. Cent. Dis. Prev. Control. 2020; published online Nov 12. Rapid Risk Assessment: Detection of new SARS-CoV-2 variants related to mink (accessed Dec 30, 2020).
9 Koopmans M. SARS-CoV-2 and the human-animal interface: outbreaks on mink farms. Lancet Infect Dis 2021; 21: 18–9.
10 Lassaunière R, Fonager J, Rasmussen M, et al. Working paper on SARS-CoV-2 spike mutations arising in Danish mink, their spread to humans and neutralization data. 2020. https://files.ssi.dk/Mink-cluster-5-short-report_AFO2.
11 Chen J, Wang R, Wang M, Wei G-W. Mutations Strengthened SARS-CoV-2 Infectivity. J Mol Biol 2020; 432: 5212–26.
12 Thomson EC, Rosen LE, Shepherd JG, et al. The circulating SARS-CoV-2 spike variant N439K maintains fitness while evading antibody-mediated immunity. Microbiology, 2020 DOI:10.1101/2020.11.04.355842.
13 Starr TN, Greaney AJ, Addetia A, et al. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. bioRxiv 2020; : 2020.11.30.405472.
14 Kemp SA, Harvey WT, Datir RP, et al. Recurrent emergence and transmission of a SARS-CoV-2 Spike deletion ΔH69/V70. bioRxiv 2020; : 2020.12.14.422555.
15 Munnink BBO, Sikkema RS, Nieuwenhuijse DF, et al. Transmission of SARS-CoV-2 on mink farms between humans and mink and back to humans. Science 2020; published online Nov 10. DOI:10.1126/science.abe5901.
16 Evolutionary annotation of SARS-CoV-2/COVID-19 genomes enabled by data from. 2020; published online Dec 23. Evolutionary annotation of SARS-CoV-2/COVID-19 genomes enabled by data from / Sergei Pond | Observable (accessed Dec 30, 2020).
17 Campbell KM, Steiner G, Wells DK, Ribas A, Kalbasi A. Prioritization of SARS-CoV-2 epitopes using a pan-HLA and global population inference approach. bioRxiv 2020; : 2020.03.30.016931.
18 Sundheds- og Ældreministeriet De fleste restriktioner lempes i Nordjylland- sum.dk. De fleste restriktioner lempes i Nordjylland | Indenrigs- og Sundhedsministeriet (accessed Dec 30, 2020).
19 Pereira F. Evolutionary dynamics of the SARS-CoV-2 ORF8 accessory gene. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis 2020; 85: 104525.
20 Zinzula L. Lost in deletion: The enigmatic ORF8 protein of SARS-CoV-2. Biochem Biophys Res Commun 2020; published online Oct 21. DOI:10.1016/j.bbrc.2020.10.045.
 Super, thank you
Super, thank you