Emergence of lineage B.1.621 in Latin America and the Caribbean
Bernardo Gutierrez1,2, Sully Márquez3, Belén Prado-Vivar3,4, Mónica Becerra-Wong3, Fernanda Zurita3, Erika Muñoz3, Juan José Guadalupe5, Leandro Patiño6, Andrés Carazco-Montalvo6, Juan Carlos Fernández-Cadena7, Derly Andrade-Molina8, Gabriel Morey-Leon9, Magaly Martinez10, Fátima Cardozo10, María Eugenia Galeano10, María Teresa Alvarez11, Oscar Rollano11, Aneth Vasquez11, Cecilia Salazar12, Ignacio Ferrés12, Paula Perbolianachis12, Mercedes Paz12, Alicia Costábile12, Pilar Moreno12, Gonzalo Moratorio12, Gregorio Iraola12, Josefina Coloma13, Patricio Rojas-Silva3, Michelle Grunauer14,15, Gabriel Trueba3, Verónica Barragán3, Paúl Cárdenas3,4*
1Departament of Zoology, University of Oxford, United Kingdom
2Universidad San Francisco de Quito, Colegio de Ciencias Biológicas y Ambientales COCIBA, Ecuador
3Universidad San Francisco de Quito, COCIBA, Instituto de Microbiología, Ecuador
4Universidad San Francisco de Quito, Centro de Bioinformática, Ecuador
5Universidad San Francisco de Quito, COCIBA, Laboratorio de Biotecnología Vegetal, Ecuador
6DTIDI, Instituto Nacional de Investigación en Salud Púbica (INSPI) Dr. Leopoldo Izquieta Pérez, Ecuador
7Laboratorio INTERLAB, Ecuador
8Universidad Espíritu Santo, Laboratorio de Omicas, Ecuador
9Universidad de Guayaquil, Ecuador
10Universidad Nacional de Asunción, Instituto de Investigaciones en Ciencias de la Salud, Paraguay
11Instituto de Investigaciones Químicas, Universidad Mayor de San Andrés, Bolivia
12Centro de Innovación en Vigilancia Epidemiológica (CiVE), Institut Pasteur Montevideo, Uruguay
13School of Public Health, University of California, Berkeley, USA
14Universidad San Francisco de Quito, COCSA, Escuela de Medicina, Ecuador
15Unidad de Cuidados Intensivos, Hospital de los Valles, Quito
Correspondence: *[email protected]
Background
Genomic surveillance of SARS-CoV-2 in various South American countries produces limited numbers of genomes mainly due to limited funding, sequencing capacity or bioinformatics and analytical expertise. However, factors like high frequencies of community transmission, low vaccination rates and weak public health systems could have contributed to the emergence of variants of interest and concern (VOIs and VOCs) like Gamma (P.1; Faria et al, 2021) and Lambda (C.37; Padilla-Rojas et al, 2021; Romero et al, 2021), all of which have shown increasing incidences in certain regions at some stage.
Additionally, the lineage B.1.621 (21H), which originated from the B.1 lineage and was first reported in Colombia (around January 2021; Laiton-Donato et al, 2021), caught our attention due to its apparently fast dissemination and its shared mutations with the Beta and Gamma variants in the spike gene (particularly amino acid substitutions N501Y and E484K); also, substitution P681H is similar (i.e. a change of a non-polar amino acid for a positively-charged one) to the Delta variant’s P681R substitution in the furin-cleavage site. Other substitutions featured in B.1.621 which are associated to changes in the binding of neutralizing antibodies to the spike protein include T95I (shared with Iota), Y144T (shared with Alpha and Eta), D950N (shared with Delta); furthermore, S: R346K, A771S, N:T205I, ORF1a:T1055A, ORF1a:T1538I, ORF1a:T3255I, ORF1a:Q3729R, ORF1 b:P314L, ORF1b:P1342S, ORF3a:Q57H, ORF3a:L106F (Hodcroft, 2021).
Since May 2021, the relative prevalence of this lineage has raised exponentially in Latin America and the Caribbean, despite its co-circulation with other VOCs and VOIs such as Alpha, Gamma, Lambda, and Iota. Also, we have observed various cases of infections caused by this lineage occurring in fully vaccinated and in reinfected patients (unpublished results by our group). From May to July, B.1.621 has been reported in countries outside of Latin America and the Caribbean, including small outbreaks in the UK, USA (Florida), Austria, Spain and France, and it has been flagged as a variant under investigation (VUI) by PHE-UK, and as a VOI by the ECDC.
Results
The presence of B.1.621 within South American countries has increased exponentially, in many locations, where it seems to outcompete Alpha, Gamma, Iota, and more recently Delta variants. The proportion of B.1.621 genomes (normalized by the sequences uploaded to GISAID per month until 31st July 2021; Fig 1) shows the rapid emergence of this variant in the continent.
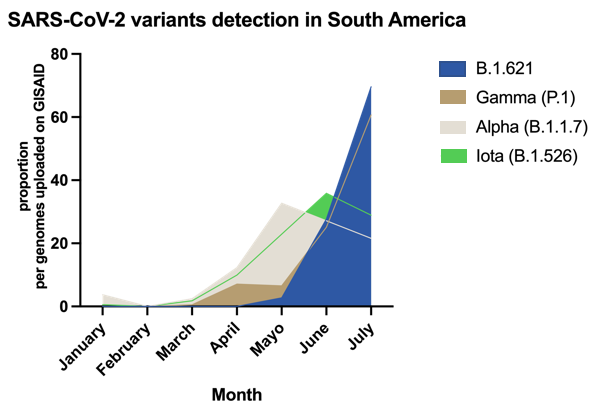
Figure 1. Normalized sequences assigned to B.1.621 in South America submitted to GISAID, plus sequences from Paraguay, Bolivia and Uruguay directly provided by the authors.
The complete GISAID B.1.621 data set (downloaded on 4th August 2021) was combined with two early B.1 sequences from Germany and Australia (EPI-ISL 450199 and EPI-ISL 509505 respectively) for tree rooting purposes and aligned using Minimap 2.17 (Li, 2018). We then constructed a maximum likelihood (ML) phylogenetic tree under a GTR+Gamma substitution model using FastTree 2.1.11 (Price et al, 2010), which estimates node support through the Shimodaira-Hasegawa (SH) approximate Likelihood Ratio test (SH-aLRT; Guindon et al, 2010). A first tree was estimated and analysed with TempEst v1.5.3 (Rambaut et al, 2016) to identify and remove outlier sequences (i.e. sequences for which the relation between their collection date and distance to the tree root are incongruent with the rest of the data set). After visually inspecting the regression and removing two outlier sequences from the USA, the tree was re-estimated and annotated by the geographic region (continent) and country where each sequence was collected. The most likely location for all tree branches were also annotated from the tips using a parsimony approach in FigTree v1.4.4. These results (Fig 2) show a cluster of sequences predominantly from North America and Europe, still identified as B.1.621 in the GISAID data base, that are basal to a well-defined lineage (SH-aLRT = 0.913) that clusters sequences predominantly from the Americas and Europe. While this clade is inferred to have emerged in North America, this is likely an artifact due to the extensive sequences available from the United States of America, as the earliest sequences that belong to the clade were reported from Colombia (Laiton-Donato et al, 2021). The lineage shows widespread circulation across Latin American countries (i.e. South and Central America and the Caribbean) as well as North America and Europe. While this tree should not be interpreted as a phylogeographic analysis (due to the wide sampling intensity gap between Europe/North America and the different countries represented from Latin America and the limitations of the parsimony approach to infer ancestral states), it does highlight the accumulating genetic diversity of this lineage and its widespread circulation across different geographic regions, which is also observable from the regression between branch lengths and sequence collection dates (Fig 1, blue box).
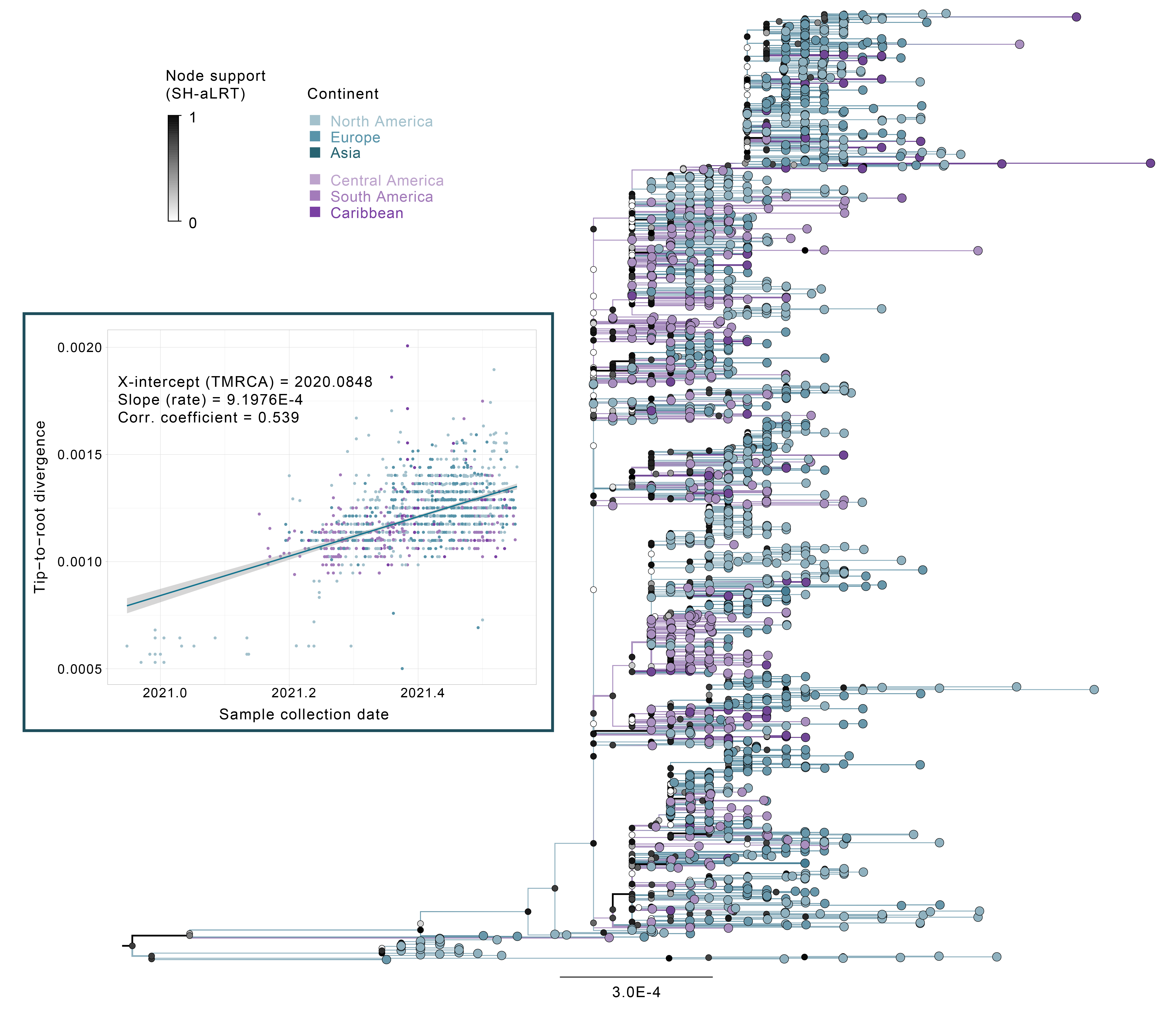
Figure 2. ML phylogenetic tree under a GTR+Gamma substitution model of GISAID B.1.621 data plus two early B.1 sequences from Germany and Australia (EPI-ISL 450199 and EPI-ISL 509505 respectively) and sequences yet to be made publicly available (Bolivia, N=1; Paraguay, N=2, Uruguay, N=5).
An analysis at a higher spatial resolution, discretised by country, shows that B.1.621 has been widely identified in Colombia (where the original lineage identification and subsequent designation request was made; Laiton-Donato et al, 2021), with multiple clusters also observed in Ecuador and other occurrences observed across countries in the Caribbean, South America and Central America. Sequences from Bolivia, Uruguay and Paraguay (highlighted in red) show multiple independent importations into each of these countries; notably, a sequence from Uruguay clusters within the B.1.621.1 lineage (associated to multiple outbreaks across Europe), suggesting a possible importation from North America or Europe rather than through circulation within Latin America. Heterogeneous sampling intensities and phylogenetic uncertainty make a finer exploration of the movements of the lineage across the region limited at this stage.
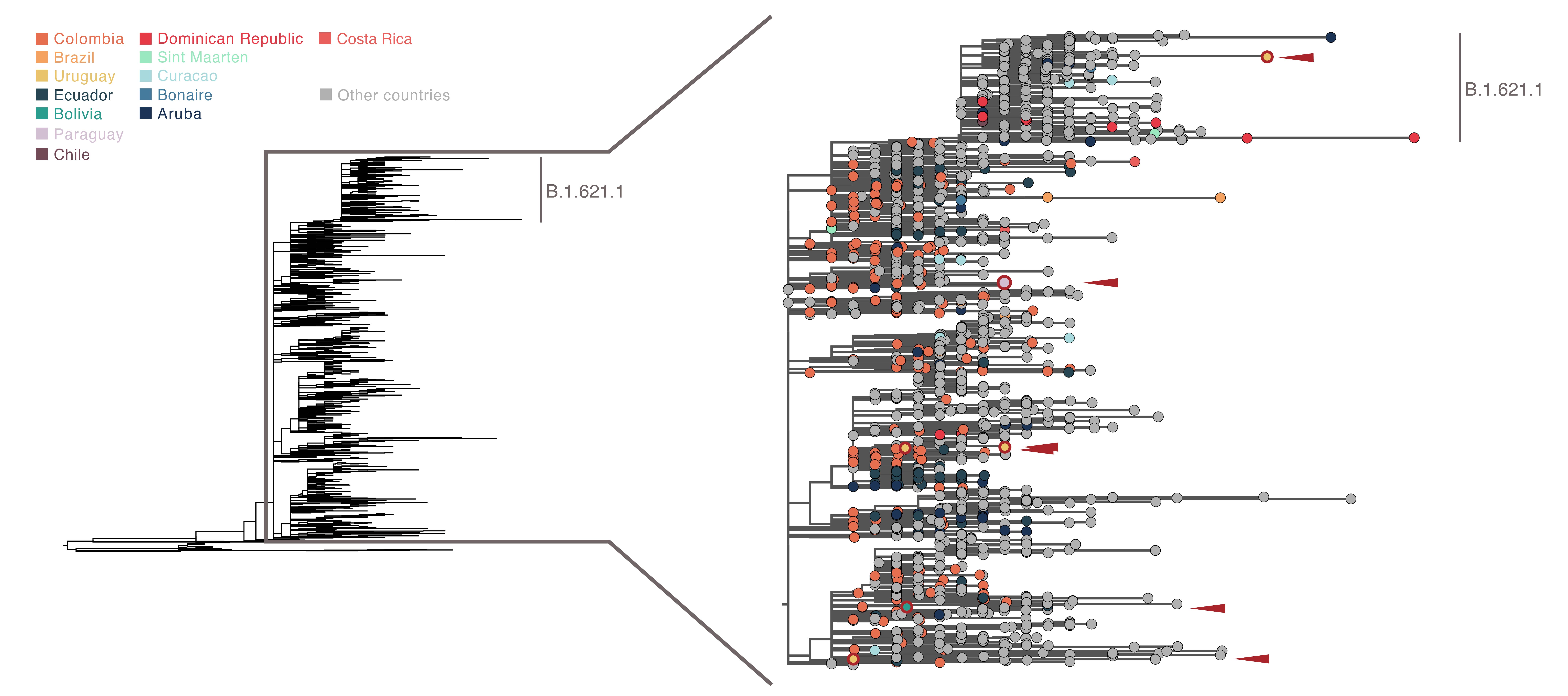
Figure 3. ML phylogenetic tree of B.1.621 (subtree from Fig 2) highlighting genomic sequences obtained from countries in the Caribbean, South America and Central America. Sequences from Bolivia, Paraguay and Uruguay are highlighted in red (their locations in the phylogeny are highlighted by red arrows).
Conclusion
The B.1.621 lineage is rapidly disseminating across Latin America and has been exported to other regions (where it has been associated with specific outbreaks), and features mutations that result in critical amino acid substitutions which have been previously observed in variants of concern (such as Alpha, Gamma and Delta); some of these key changes (such as N501Y and E484K, shared with the Beta variant) have been associated with immune-evasion and improved cell invasion (Xie et al, 2021; Zhou et al, 2021; Jangra et al, 2021). It should be widely recognized as a VOI, as has been proposed by Public Health England in the UK (as VUI-21JUL-01) and the ECDC, and its spread should be closely monitored in the Americas and elsewhere. While it is hard to assess the expansion of a lineage in the region from sparsely sampled genomic data (as is the case for many countries in Latin America and the Caribbean), its increasing frequency over time among the reported genomic sequences should be taken as evidence of its local expansion in spite of its co-circulation with other variants with demonstrated increased transmissibility. Further analyses exploring its transmissibility, viral fitness and drivers of spatial transmission could provide additional insights into the potential reach of the variant in Latin America and beyond.
Acknowledgements
This work is part of the “Red para el desarrollo de instrumentos innovadores aplicados a la investigación epidemiológica en América del Sur”. We also want to acknowledge the Grupo de Trabajo Interinstitucional de Vigilancia Genómica de SARS-CoV-2 (Uruguay), the Ecuador Clinical-COVID19 Consortium and the various groups who have shared B.1.621 genomic sequences through GISAID, particularly from Colombia where the lineage was first described.
References
Faria NR, Mellan TA, Whittaker C, Claro IM, da Silva Candido D et al. 2021. Genomic and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 21(372): 815-821.
Padilla-Rojas C, Jimenez-Vasquez V, Hurtado V, Mestanza O, Molina IS et al. 2021. Genomic analysis reveals a rapid spread and predominance of Lambda (C.37) SARS-COV-2 lineage in Peru despite circulation of variants of concern. J Med Virol. Accepted Author Manuscript. https://doi.org/10.1002/jmv.27261
Romero PE, Dávila-Barclay A, Salvatierra G, González L, Cuicapuza D et al. 2021. The emergence of SARS-CoV-2 variant Lambda (C.37) in South America. medRxiv,https://doi.org/10.1101/2021.06.26.21259487
Laiton-Donato K, Franco-Muñoz C, Álvarez-Díaz DA, Ruiz-Moreno HA, Usme-Ciro JA et al. 2021. Characterization of the emerging B.1.621 variant of interest of SARS-CoV-2. medRxiv, https://doi.org/10.1101/2021.05.08.21256619
Hodcroft EB. 2021. “CoVariants: SARS-CoV-2 Mutations and Variants of Interest." https://covariants.org/
Li H. 2018. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 34: 3094–100.
Price MN, Dehal PS, Arkin AP. 2010. FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 5(3): e9490.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Systematic Biology 59(3): 307–321.
Rambaut A, Lam TT, Carvalho LM, Pybus OG. 2016. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evolution 2(1): vew007.
Xie X, Liu Y, Liu J, Zhang X, Zou J et al. 2021. Neutralization of SARS-CoV-2 spike 69/70 deletion, E484K and N501Y variants by BNT162b2 vaccine-elicited sera. Nat Med 27: 620–621.
Zhou D, Dejnirattisai W, Supasa P, Lui C, Mentzer AJ et al. 2021. Evidence of escape of SARS-CoV-2 variant B.1.351 from natural and vaccine-induced sera. Cell 189: 2348-2361.
Jangra S, Ye C, Rathnasinghe R, Stadlbauer D; Personalized Virology Initiative study group, Krammer F, Simon V, Martinez-Sobrido L, García-Sastre A, Schotsaert M. 2021. SARS-CoV-2 spike E484K mutation reduces antibody neutralisation. Lancet Microbe 2(7): e283-e284.